Our tool is under active development and feedback is very much appreciated.
learnMSA2: deep protein multiple alignments with large language and hidden Markov models
![]()
- Aligns large numbers of protein sequences with above state-of-the-art accuracy
- Can utilize protein language models (
--use_language_model) for significantly improved accuracy - Enables ultra-large alignment of millions of sequences
- GPU acceleration, multi-GPU support
- Scales linear in the number of sequences (does not require a guide tree)
- Memory efficient (depending on sequence length, aligning millions of sequences on a laptop is possible)
- Visualize a profile HMM or a sequence logo of the consensus motif
- Requires many sequences (in most cases starting at 1000, a few 100 might still be enough) to achieve high accuracy
- Only for protein sequences
- Increasingly slow for long proteins with a length > 1000 residues
Recommended ways to install learnMSA along with Tensorflow + GPU:
We provide a hassle-free docker image including GPU and pLM support.
singularity build learnmsa.sif docker://felbecker/learnmsa:2.0.9
singularity run --nv learnmsa.sif learnMSA
Running the container with --nv is required for GPU support.
- Create a conda environment:
conda create -n learnMSA python=3.12
conda activate learnMSA
- Install learnMSA (cuda toolkit included):
pip install learnMSA
- Additional installs for sequence weights (recommended!):
conda install -c bioconda mmseqs2
You may have to set up Bioconda channels.
- Additional installs for language model support (recommended!):
pip install torch==2.2.1 tf-keras==2.17.0
- (optional) Verify that TensorFlow and learnMSA are correctly installed:
python3 -c "import tensorflow as tf; print(tf.__version__, tf.config.list_physical_devices('GPU'))"
learnMSA -h
Recommended way to align proteins:
learnMSA -i INPUT_FILE -o OUTPUT_FILE --use_language_model --sequence_weights
Without language model support (recommended for speed and for proteins with very high sequence similarity):
learnMSA -i INPUT_FILE -o OUTPUT_FILE --sequence_weights
Note: If you installed learnMSA via docker/singularity, you have to run singularity run --nv learnmsa.sif learnMSA -i ...
We always recommend to run learnMSA with the --sequence_weights flag to improve accuracy. This requires mmseqs2 to be installed (see above). Sequence weights (and thus the mmseqs2 requirement) are turned off by default.
To output a pdf with a sequence logo alongside the msa, use --logo. For a fun gif that visualizes the training process, you can use --logo_gif (attention, slows down training and should not be used for real alignments).
Run the notebooks learnMSA_demo.ipynb or learnMSA_with_language_model_demo.ipynb with juypter.
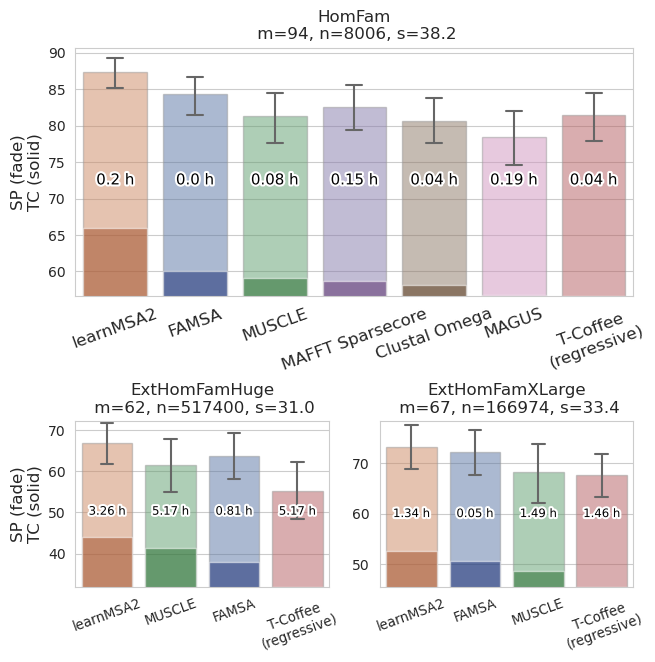
Becker F, Stanke M. learnMSA2: deep protein multiple alignments with large language and hidden Markov models. Bioinformatics. 2024
Becker F, Stanke M. learnMSA: learning and aligning large protein families. GigaScience. 2022