Template matching
Features in a tomogram that resemble a structural 'template' can be localized in an automated fashion using 'template matching'. In this approach a 3D template is correlated with a given tomogram. In this procedure the different possible rotations and translations are sampled exhaustively using the algorithm described in Förster et al, Meth. Enzymol. 483:215-43 (2010).
Here we describe how to set-up template matching--both from the GUI and from the command line. Additionally, we also describe how to get the best performance from template matching in the GUI. For command line instructions, scroll to the bottom.
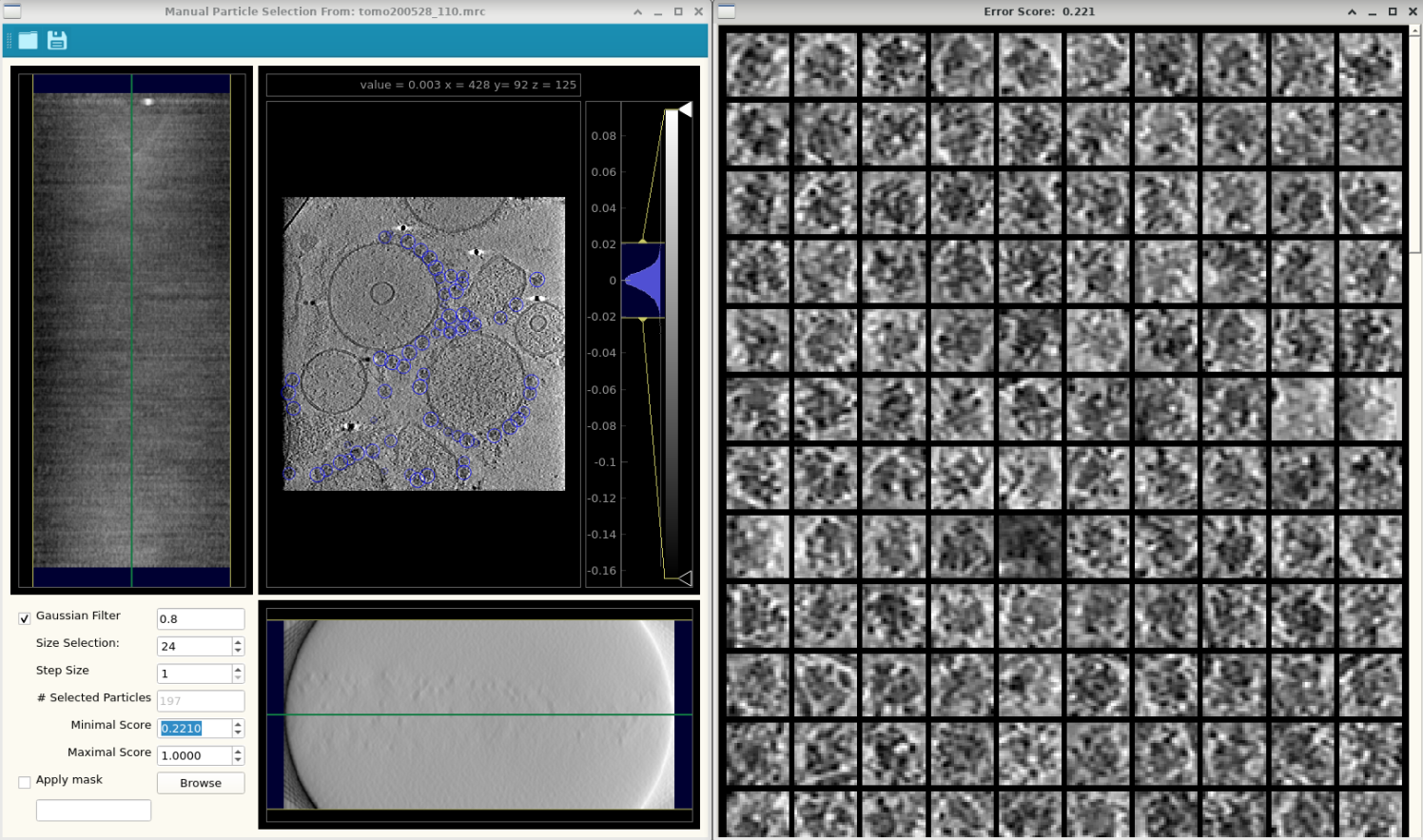
figure: Ribosome template matching results on ER-derived vesicles viewed in the pytom particle picking GUI. (data from Gemmer et al., 2023)
NOTE: For easy execution, the tomograms (mrc files) need to be stored in a single directory. This can be the reconstruction folder from Warp. Or the tomogram directory in your pytom project.
The easiest way to start template matching is from the Batch Template Match tab. Multiple jobs can be set-up simultaneously.
Go to: Particle Picking -> Template Matching -> Batch Template Match
After pressing Refresh, in the first pop-up browse to the folder where the tomograms are stored and select it. Then in the next two pop-ups, browse to the template location, double-click the template file and press OK, then do the same for the mask. You can also double click multiple templates/masks and later on select the one you want to run with.
Jobs are activated per tomogram by clicking either or both, the Run and Mirrored check boxes. Mirorred will
run the job with a mirrored version of the template. This depends on how the particles are oriented in your tomogram.
You can run both versions and plot correlation scores against each other to find the handedness (more info on this
in the PyTomGUI Tutorial and also below).
For all parameters note that modifying the top row of the table allows to simultaneously set a value for all tomograms.
-
Spherical maskis a true/false option that specifies whether or not the mask is spherical. In case its spherical always set this to true, it will greatly speed up the calculation! Because of this speed-up, always try to use a spherical mask (especially for ribosomes). For more difficult particle searches non-spherical mask can become important, but can also introduces bias. -
Wedge Angle 1the first angle of the missing wedge. Found by subtracting the largest negative angle from 90. I.e. a tilt-series from -54 to +51, gets a first wedge angle of 90 – 54 = 36 degrees. -
Wedge Angle 2the second angle of the missing wedge. Found by subtracting the largest positive angle from 90. I. e. a tilt-series from -54 to +51, gets a second wedge angle of 90 – 51 = 39 degrees. -
Angle listsets the rotational search of the template. PyTom comes with preset angular searches that you can choose from. Each search includes the angular increment and the number of orientations that are sampled for it:angles_12.85_7112.emindicates an ~13 degrees increment with 7112 rotations of the template. The optimal search for your template is found from the Crowther criterion$$\Delta \alpha = \frac{180}{\pi r_{max} d}$$ , where$r_{max}$ is the max resolution in fourier space of either template or tomogram, and d is the particle diameter. A ribosome (d=290A) in a 20A tomogram ($r_{max}$ is$\frac{1}{2*20} A^{-1}$ ), has an optimal search of 8 degrees. We can choose for the optimal search from the closest available angle lists, which would here beangles_7_45123.em, i.e. half a ~50,000 rotations. Optionally you can oversample, which has some benefits, but it will take more time. -
Start ZandEnd Zallow to limit the search range to a specific z range of the tomogram. Often the sample layer does not occupy the full z height of the tomogram. Limiting the search area speeds up calculation. These parameters denote the start and end index of the area to search. -
x split,y split, andz splitset splitting of the tomogram over multiple processes to allow parallel processing. The splits are multiplied with each other to find the total number of subvolumes (and processes) needed. For example, setting xsplit=2 and ysplit=2 results in 4 boxes, while setting xplit=2 and ysplit=4, results in 8 boxes. You will need to assign processes to subvolume (more on that below!). For running template matching on CPU’s you will need quite some processes to achieve feasible runtime. Generally, we advise 16 processes which can be achieved by an xplit,ysplit,zsplit=4,4,1 . -
GPU ID, one or multiple gpu indices (separated by commas) to run the template matching on. With an xplit,ysplit,zsplit=2,2,1, we need to assign a gpu to each subvolume. Setting gpu’s to 0,1,2,3 will enable the right amount of gpu’s to run the job. Larger tomograms will need to be split over multiple gpu’s to allow the tomogram to fit in memory. Running the subvolumes sequentially on a single gpu is currently not implemented (sadge). - Below the table you can set the number of nodes to submit to. For submission to a gpuq just set it to 1. Also activate the queue check box if you are submitting the job.
IMPORTANT For both CPU and GPU processes the job settings need to be specified! To do this, go to the settings menu and select BatchTemplateMatch from the Job Submission Parameters-dropdown menu. You might need to set the Submit to: depending on gpu/cpu submission. On our cluster we have a separate gpu and cpu queue in slurm: gpuq, and defq. Node can likely be set to 1 if a single node offers enough cores for the job. Cores need to be set equal to the number of processes. For the 4 gpu job above you will need to assign 4 cores (always need to match with number of gpus), while for the 16 subvolume cpu job you need to specify 16 cores here. When running a job locally (no submission to slurm), the cores are also read from here, so you still need to specify it.
This is currently a bit limited. Pytom will inform you on the bottom of the GUI whether you have an actively running template matching job, but it wont inform you on the number of tomograms finished. The only way is to open the Queue (stack) icon from the top toolbar of the GUI. There you can switch between Local (non-submitted) and Queued jobs. Each running job has options to show the job file, open the log, or terminate. For template matching opening the log file can indicate you some progress. All tomogram output is pasted underneath each other for a batch job. At the end of a run on a tomogram the runtime in seconds is written (search for ‘The overall execution time: …’). Based on total runtime and the time per tomogram you can estimate how long it will take.
Each template matching job will write out a score and angle volume. The score volume stores the highest correlation score for each voxel and the angle volume the corresponding angle index of the highest score. From these volumes we can extract candidates with different settings. This prevents have to rerun the precious template matching.
In Template Matching -> Batch Extract we can run it for all tomograms simultaneously (after template matching has finished). First refresh the tab to load all job outputs, you can select which jobs to put in the table (but automatically all jobs will be listed).
- Check the box on the top row (Apply to all) to run on all jobs
- Output name and subtomogram directory can be set if required, but you need a unique name for each particle list. The default particle list name is found based on tomogram name and template name and is recommended to leave as is.
-
Radiusis the most important parameter here! Peaks are extracted sequentially, starting from the highest correlation score. After a peak is extracted the area will be masked depending on this radius. This is important because we don’t want to extract multiple copies of a particle. The radius should be set depending on the size of the particle. A 290A ribosome will encompass 14.5 pixels in a 20A tomogram (290/20). Thus our extraction radius should be 7 or 8 pixels. -
# Candidatessets how many peaks to extract in total. Depends on the number of particles that are extracted per tomogram. If you want to do any true positive estimation (see below), this needs to be increased to include sufficient false positives: double the number of expected true positives to get a good extraction. -
Min. Scoresets the minimum score of extracted peaks. If we attempt to extract 500 candidates, but at #42 the score drops below the minimum score, pytom will stop extracting.
Candidate extraction can only be run on single CPUs. To get the most efficiency, the resources per extraction need to be limited. In the GUI job settings menu, go to Job Submission Parameters for BatchExtractCandidates and limit the resources to 1 node and 1 core! When starting the job, increase the number of nodes at the bottom of the batch table to the number of processes you want to run parallel. Setting it to 20 will run at most 20 candidate extractions in parallel!
True positives in the template matching results can be estimated to find a correlation score threshold. This can be applied during candidate extraction to give some classification to the template matching results. It can also give inside into the handedness of the tomogram. There is two ways to do this: comparing scores between mirrored versions of the template and estimation via Gaussian fitting.
- Mirroring the template to find a score threshold. The idea here is to run template matching with the regular and mirrored orientation and plot the scores against each other. The higher scoring orientation is assumed to show the handedness of the tomogram. In the plot menu of the GUI toolbar you can plot two particle list against each other. For more detailed info see the PyTomGUI Tutorial.
- Estimating true positives via Gaussian fitting. From the command line you can run the
plotGaussianFit.pyscript which attempts to fit a distribution to the true positives in your results. You need to give an input xml particle list (-f), a number of bins for the histogram (-n; ~25 works well), initial guess of the gaussian peak location (-p; 25 // 2 ~= 12 works fine), and use--cropPlot. The script will print some output and write a png file with results (same name as particle list). Fitting is not super robust so you might need to play around with the parameters. For accurate fitting it might be effective to pool particle list of a few tomograms (viaCreate Particle List->Batch). Below an example of a good fit!

figure: Gaussian fit to extracted correlation scores from template matching. The positive and negative results can be modelled as a bimodal model of two gaussians (blue line). The red line indicates a fit for the true positives. The dotted horizontal line indicates the optimal score cutoff based on the ROC curve on the right (green point). The ROC curve is estimated from the bimodal model. The closer the AUC (area under curve) value is to 1, the better the classifier is.
Template matching needs to be run in three stages: (1) generating a xml job file with all execution parameters (keeps track of you work nicely!), (2) running the job with varying parallel processing options, and (3) extracting candidates. Explanation for the parameters can be read in the GUI template matching setup (see above!).
The localizationJob.py script allows you to create the xml file by prompting to fill in the appropriate parameters.
The volume (tomogram), reference, and mask can all be mrc or em files.
NAME
localizationJob.py
DESCRIPTION
Create a localization job. Documentation is available at
http://www.pytom.org/doc/pytom/localization.html
OPTIONS
-v, --volume Volume : the big volume (Is optional: No; Requires arguments: Yes)
-r, --reference Reference : the molecule searched (Is optional: No; Requires arguments: Yes)
-m, --mask Mask : a mask (Is optional: No; Requires arguments: Yes)
--spherical_mask Set this option if the mask is spherical to speed up the calculation. (Is optional: Yes; Requires arguments: No)
--wedge1 Wedge : first tilt angle. Must be 90-tilt! (Is optional: No; Requires arguments: Yes)
--wedge2 Wedge : second tilt angle. Must be 90-tilt! (Is optional: No; Requires arguments: Yes)
-a, --angles Angles : name of angle list. Either :
angles_50_100.em
angles_38.53_256.em
angles_35.76_320.em
angles_25.25_980.em
angles_19.95_1944.em
angles_18_3040.em
angles_12.85_7112.em
angles_11_15192.em
angles_07_45123.em
angles_3_553680.em
(Is optional: No; Requires arguments: Yes)
-d, --destination Destination : destination directory (Is optional: No; Requires arguments: Yes)
--zstart Start of z height subregion to search. (Is optional: Yes; Requires arguments: Yes)
--zend Start of z height subregion to search. (Is optional: Yes; Requires arguments: Yes)
-b, --band Lowpass filter : band - in pixels (Is optional: Yes; Requires arguments: Yes)
-j, --jobName Specify job.xml filename (Is optional: No; Requires arguments: Yes)
-h, --help Help. (Is optional: Yes; Requires arguments: No)
AUTHORS
Thomas Hrabe
The result of this script will be a job.xml and a job.sh file.
<JobDescription Destination="./results/">
<Volume Filename="./tomogram.em"/>
<Reference Weighting="" File="./reference.em"/>
<Mask Filename="./mask.em" Binning="1" isSphere="True"/>
<WedgeInfo Angle1="30" Angle2="30" CutoffRadius="0.0" TiltAxis="custom">
<Rotation Z1="0.0" Z2="0.0" X="0.0"/>
</WedgeInfo>
<Angles Type="FromEMFile" File="angles_12.85_7112.em">
<RefinementParameters Shells="6.0" Increment="10.0"/>
<OldRotations/>
</Angles>
<Score Type="FLCFScore" Value="-100000000">
<DistanceFunction Deviation="0.0" Mean="0.0" Filename=""/>
</Score>
</JobDescription>
Please note that a subregion of the tomogram can be defined, in order to reduce the number of false positives, and speeding up the calculation. The first three numbers of the series describe the starting coordinates of the subregion (x-coord, y-coord, and z-coord). The second three numbers indicate the size of subregion in voxels (x-dim, y-dim, and z-dim). If (0,0,0,0,0,0) is supplied, no subregion is selected.
The script localization.py allows computing the constrained local correlation of a tomogram and a reference in a
local area described by a mask. The script supports MPI, which allows running the script on large computer clusters.
Calling the script for GPU template matching looks like this (volume split in 4 subvolumes and ran on 4 gpu's):
mpiexec --tag-output -n 4 localization.py -j job.xml -x 2 -y 2 -z 1 --gpuID 0,1,2,3
Running on 16 cpu's (while the volume is split in 16 parts:
mpiexec --tag-output -n 16 localization.py -j job.xml -x 4 -y 4 -z 1
The correlation volume and corresponding orientations generated above need to be interpreted. The script
extractCandidates.py simply determines the peaks of the correlation volume and the corresponding orientations that are all stored in a particle list (xml file).
extractCandidates.py -j job.xml -n 500 -s 7 -r scores.em -o angles.em -p pl.xml -v 0.1 -t subtomogram_folder
For usage, you need to specify the correlation (score) volume, where peaks are located -r score.em , and volume
with best angle indexes -o angles.em. This will generate a particle list -p pl.xml in the current directory. The
particle list will contain -n 500 particles or until their minimum score drops below 0.1 -v 0.1. The cut-out radius
around each peak is -s 7. In the resulting particle list, all particles will have a prefix determined by the -t
option. Please note that you can include the complete (absolute) path to the particles, here, too.
Some other notes on parameters:
- Extract candidates can also write out an old school motl file with the
-moption. - For non-spherical template matching you can also specify a structured mask for cutting out space around extracted
candidates (instead of the sphere that is cut out with the
-soption). - You can provide an edge
--marginto not extract particles that are close to the edges of the tomogram. - You can provide a
--tomogram-maskto only extract candidates in certain regions of the tomogram, such as vesicles or specific organelles. The mask will need to be generated in other software and its essential that it matches the dimensions of the scores.em and angles.em volumes exactly. More information on this in Vesicle/organelle masking.