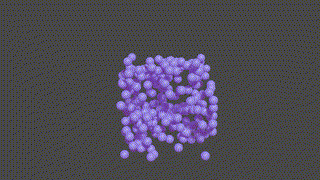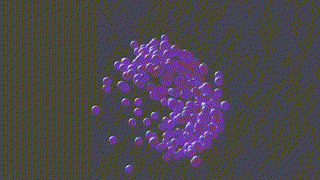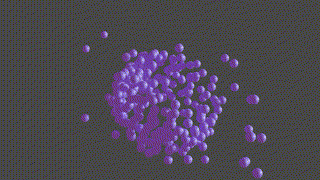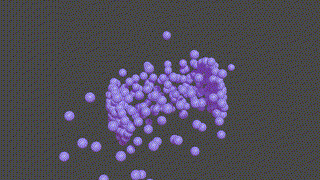

A PyTorch based neural network program designed to recontruct protein complexes in 3D from a series of 2D microscopy images. We predict the pose and structure using a Convolutional Neural Network.
HOLLy takes a number of images (it can also generate images from a ground-truth point-cloud) and trains on these attempting to improve it's own internal representation of what it thinks the ground-truth object is. At the end of training, an obj or *ply file will be produced, representing the object the network converged upon.
HOLLy is split into train.py and run.py with the actual net stored in in net/net.py. eval.py will evaluate a trained network, creating statistics and visualistions.
net/renderer.py contains the code for the differentiable renderer. data/loader.py, along with data/buffer.py and data/batcher.py create our simulated data for all the tests, including adding noise. data/imageload.py is similar, but for the real microscopy images.
(The diagram below is animated and may take a little time to appear).

This readme covers most of the details of HOLLy, but we also have more documentation available at Read The Docs.
Requirements include:
- A Linux system setup to perform deeplearning, with appropriate nvidia drivers installed.
- Python version 3
- CUDA setup and running
- A GPU with at least 4G of memory (more if you are fitting more points).
And one of the following
If you want to generate lots of statistics for use later you'll also need all of the following installed and running. Initially, you shouldn't need these but you might see a few warnings pop up:
If you want some fancy formatted results or if you are running the tl;dr script, install the following:
Finally, in order to view the model the network has derived, you'll need a program capable of displaying obj or ply files. These are pretty numerous, but here are a few we use:
- meshlab
- [blender(https://www.blender.org/)
Chances are, if you are running a Linux distribution, you will have these already and if not, they'll be in your repository management system (like apt or pacman).
All of the python requirements are listed in the requirements.txt file (there aren't too many).
Run the script tldr.sh from the top directory. You will need miniconda installed and a computer running Linux, capable of trainin a large neural network with Pytorch. As a guide, a machine with several cores, >=16G memory and an nVida GPU such as a GTX 2080Ti will work reasonably well.
The script creates a miniconda environment called "holly", downloads packages and starts running an experiment, followed by a generation of results and a final.ply file representing the learned model. Once this script has completed, take a look at the Outputs section of this readme to understand what the network has produced.
Assuming you have installed miniconda, start with the following:
conda create -n holly python
conda activate holly
pip install -r requirements.txt
There is a dockerfile included in docker/holly that will create a container that you can use to train our network.
Assuming you have docker installed, you can create the docker container like so:
docker build -t holly docker/holly
We can simulate data from a wavefront obj file, several of which are included with this repository. Alternatively, you can provide your own.
To train a new network on simulated data, run the following command in the top level of the project, assuming you are inside the miniconda environment:
python train.py --obj objs/teapot_large.obj --train-size 40000 --lr 0.0004 --savedir /tmp/test_run --num-points 230 --epochs 20 --sigma-file run/sigma_quick.csv
It is also possible to use the provided bash script to execute training in a given directory, saving the various options and code versions to various files. This is more useful if you want to run many tests, saving the various settings.
mkdir ../runs/temp
cd run
cp run.conf.example ../../runs/temp/run.conf
./train.sh ../../runs/temp "My first run"
The bash script train.sh looks for a file called run.conf that contains the required data for training a network. The file run.conf.example can be copied to run.conf. Make sure the directory referred to for saving the results exists.
Using docker, one can run the same command as follows:
mkdir experiment
docker run --gpus=all --volume="$PWD:/app" holly python train.py --obj objs/teapot_large.obj --train-size 40000 --lr 0.0004 --savedir /tmp/experiment --num-points 230 --epochs 20 --sigma-file run/sigma_quick.csv
Confirm that docker can see the gpu: docker run --gpus all holly nvidia-smi
Your GPU should be listed.
Docker needs the nvidia-docker2 package in order to access gpus. On Ubuntu 20.04 LTS one needs to run the following commands:
curl -s -L https://nvidia.github.io/nvidia-docker/gpgkey | apt-key add -
curl -s -L https://nvidia.github.io/nvidia-docker/ubuntu20.04/nvidia-docker.list > /etc/apt/sources.list.d/nvidia-docker.list
apt update
apt -y install nvidia-docker2
systemctl restart docker
There are a large number of options you can pass to the network. These are listed in the train.py file. The most important are listed below:
- --save-stats - save on-going train and test statistics.
- --obj - when simulating data, this is the path to the object file we want to use.
- --train-size - the size of the training set
- --lr - the learning rate
- --num-points - how many points do we want to try and fit to the structure.
- --epochs - how many epochs should we run for?
- --sigma-file - the path to the file containing the per-epoch sigmas.
- --image-width - the width of the input and output images
- --image-height - the height of the input and output images
Training usually takes around 4 hours on a nVidia 2080Ti system when running for 20 epochs on an 80,000 size training set.
The final structure discerned by the network is saved as both an obj and a ply file. Both can be viewed in a program like Meshlab or Blender. The files can be found in:
<save_directory>/plys/shape_e<epoch>_s<step>.ply
<save_directory>/objs/shape_e<epoch>_s<step>.obj
To use real, experimental data one must have a number of images, rendered using a particular sigma. These images need to be held within a directory stucture that matches the sigma file passed in. For example:
images
|___10.0
|____image0.fits
|____image1.fits
|___8
|____image0.fits
|____image1.fits
|___1.2
|____image0.fits
|____image1.fits
The sigma file (sigma_images.csv) would be: 10,8,1.2
Images should be in FITS Format (they support floating point values). We would then run the following command: python train.py --fitspath images --train-size 80000 --lr 0.0004 --savedir /tmp/runs/test_run --num-points 230 --no-translation --epochs 20 --sigma-file sigma_images.csv
A test data set is available for download from Zenodo. This is a big download - just under 50G! However it does have all the data ready for use, pre-rendered.
Once you've donwloaded and unzipped the data, place all the images into a directory structure like this:
holly
|___run
|___paper
|____10.0
|____8
etc
...
Use the script run_cp.sh in the run directory. This assumes you've installed the environment with either miniconda or docker. This script assumes you have downloaded the dataset and placed it the correct directory.
If you want to see the results of a real CEP152 run - the ones from our paper - you can download them from Zenodo too!
The data used in the paper comes from Suliana Manley's research group and is available as a MATLAB/HDF5 file. In order to use it with this network, you will need to render the data to a series of FITs images. We have written a program to do this called CEPrender. You can download and install this program, then generate the data as follows:
cargo run --release --bin render /data/Cep152_all.mat /paper/10 24 10 10 cep_all_accepted.txt
cargo run --release --bin render /data/Cep152_all.mat /paper/9 24 9 10 cep_all_accepted.txt
...
..
.
cargo run --release --bin render /data/Cep152_all.mat /paper/1.41 24 1.41 10 cep_all_accepted.txt
... filling the missing steps with the other sigma levels. This takes quite a while, even with only 10 augmentations. The pre-rendered data can be found on Zenodo complete the with the full instructions for generating it.
The estimated energy use to train a simulated model is 2.1kWh based on a measurement of 623.4kWh over 166 days. In this period, 298 models were trained and evaluated. This was confirmed by cross-checking against the wattage of the GPU and the time spent to generate an average model.
Once you have a trained network, you can generate the final outputs using the generate_stats.sh script found in the run directory.
./generate_stats.sh -i <path to your results directory> -g <path to object file> -a -z
So with the test example given above:
./generate_stats.sh -i /tmp/runs/test_run -g ../objs/teapot.obj -a -z
This relies on having imagemagick 'montage' command and the correct python libs installed.
This script loads the network and tests it against the ground_truth (if we are using simulated data)
Due to the nature of this work, we used Jupyter Notebooks, Redis and PostgreSQL to save our various statistics, using the code found in stats/stats.py. By default, these statistics include:
- Train and test errors.
- Input translations and rotations that make up the dataset.
- Output translations and rotations the network decided upon.
- Input and output sigma.
The training machine must be running an instance of either Redis or PostgreSQL. If both are running, the program will use both. Imagemagick and ffmpeg are used to create montage images and animations.
At some point in the future, we might move to Tensorboard and / or Weights and Biases.
Our code comes with a considerable number of tests. These can be run from the top level directory as follows:
python test.py
Individual tests can be run as follows:
python -m unittest test.data.Data.test_wobble
The CEP152 data we used in the paper with HOLLy can be found on Zenodo at https://zenodo.org/record/4751057 and the results we mention in the paper can also be downloaded from Zenodo at https://zenodo.org/record/4836173.
When running train.py, there are a number of options one can choose.
--batch-size
Input batch size for training (default: 20).
--epochs
Number of epochs to train (default: 10).
--lr
Learning rate (default: 0.0004).
--spawn-rate
Probabilty of spawning a point (default: 1.0).
--max-trans
The scalar on the translation we generate and predict (default: 0.1).
--max-spawn
How many flurophores are spawned total (default: 1).
--save-stats
Save the stats of the training for later graphing.
--no-cuda
Disables CUDA training.
--deterministic
Run deterministically.
--normalise-basic
Normalise with torch basic intensity divide.
--seed
Random seed (default: 1).
--cont
Continuous sigma values
--log-interval
How many batches to wait before logging training status (default 100)
--num-points
How many points to optimise (default 200).
--aug
Do we augment the data with XY rotation (default False)?
--num-aug
How many augmentations to perform per datum (default 10).
--save-interval
How many batches to wait before saving (default 1000).
--load
A checkpoint file to load in order to continue training.
--savename
The name for checkpoint save file.
--savedir
The name for checkpoint save directory.
--allocfile
An optional data order allocation file.
--sigma-file
Optional file for the sigma blur dropoff.
--dropout
When coupled with objpath, what is the chance of a point being dropped? (default 0.0)
--wobble
Distance to wobble our fluorophores (default 0.0)
--fitspath
Path to a directory of FITS files.
--objpath
Path to the obj for generating data
--train-size
The size of the training set (default: 50000)
--image-height
The height of the images involved (default: 128).
--image-width
The width of the images involved (default: 128).
--test-size
The size of the training set (default: 200)
--valid-size
The size of the training set (default: 200)
--buffer-size
How big is the buffer in images? (default: 40000)
3D Structure from 2D Microscopy images using Deep Learning - Frontiers in Bioinformatics
@article{blundell3DStructure2D01,
title = {{{3D Structure}} from {{2D Microscopy}} Images Using {{Deep Learning}}},
author = {Blundell, Benjamin James and Rosten, Ed and Ch'ng, QueeLim and Cox, Susan and Manley, Suliana and Sieben, Christian},
year = {2021},
journal = {Frontiers in Bioinformatics},
volume = {0},
publisher = {{Frontiers}},
issn = {2673-7647},
doi = {10.3389/fbinf.2021.740342},
copyright = {All rights reserved},
langid = {english},
}
See the CITATION.cff file.
You can contribute to this project by submitting a pull request through github. Suggestions and feedback are welcomed through Github's Issue tracker. If you want to contact the authors please email Benjamin Blundell at [email protected].
- Docker image based on - https://github.com/anibali/docker-pytorch
"And the moral of the story; appreciate what you've got, because basically, I'm fantastic!" - Holly. Red Dwarf