-
Notifications
You must be signed in to change notification settings - Fork 0
/
Part1.Rmd
304 lines (201 loc) · 11.3 KB
/
Part1.Rmd
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
269
270
271
272
273
274
275
276
277
278
279
280
281
282
283
284
285
286
287
288
289
290
291
292
293
294
295
296
297
298
299
300
301
302
303
---
title: "R Introduction for Biology"
author: "Mark Dunning"
date: '`r format(Sys.time(), "Last modified: %d %b %Y")`'
output:
html_notebook:
toc: yes
toc_float: yes
css: stylesheets/styles.css
---
```{r setup, include=FALSE,message=FALSE,warning=FALSE}
knitr::opts_chunk$set(echo = TRUE,message=FALSE)
```
# Dealing with data
The [***tidyverse***](https://www.tidyverse.org/) is an eco-system of packages that provides a consistent, intuitive system for data manipulation and visualisation in R.
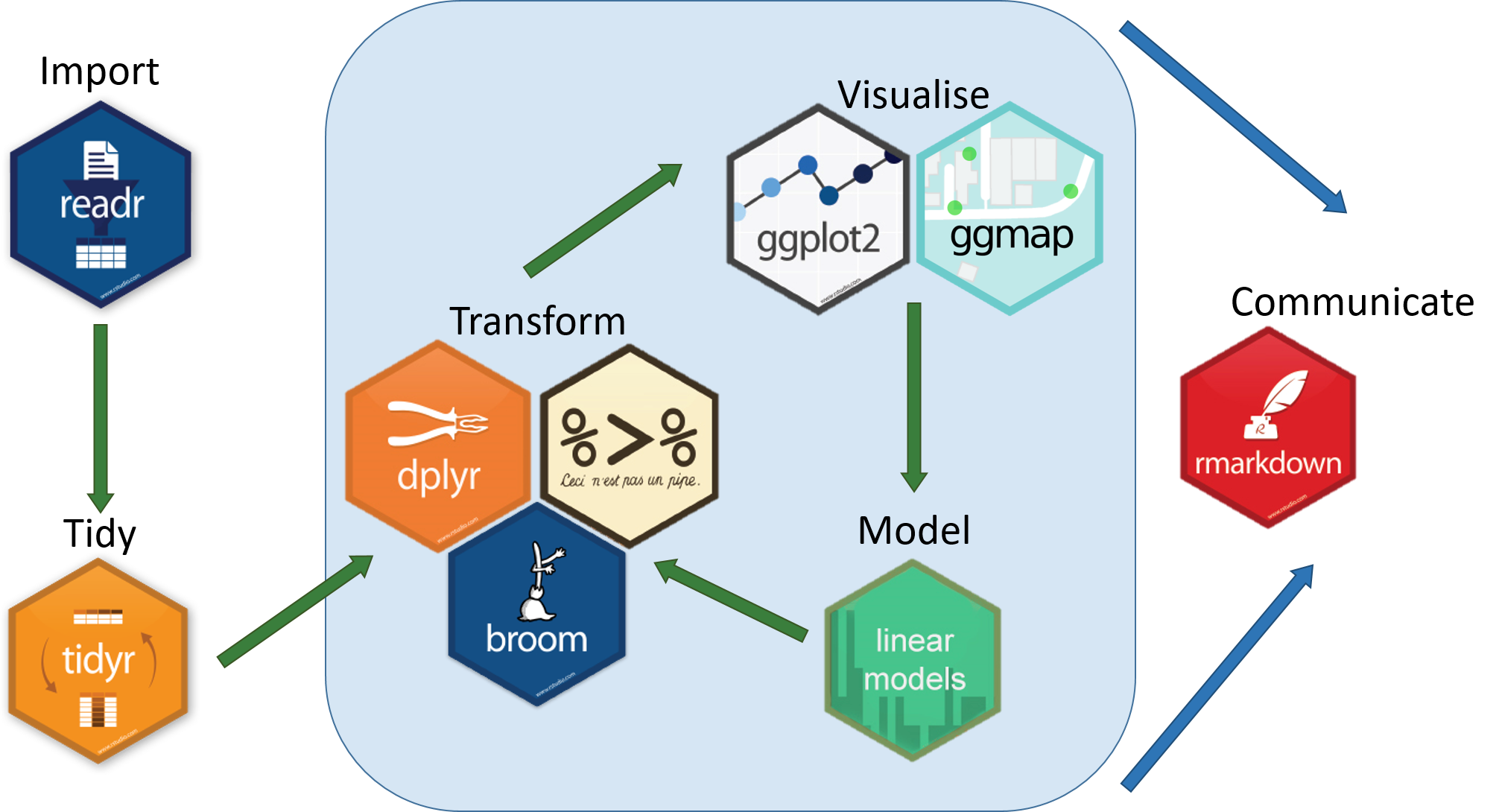
A spreadsheet that we would normally analyse in Excel can can be imported into R by using a *function* from the readr package.
- `read_csv`
- `read_tsv`
The readxl package can also import files in Excel's own format
- `read_xls`
- `read_xlsx`
We will use the file `tcga_clinical_CLEANED.tsv` as an example. This file contains information about biological samples that were included as part of the TCGA (The Cancer Genome Atlas) project - all tumour types.
As the file name ends in `.tsv` the function we want is probably `read_tsv`
```{r warning=FALSE,message=FALSE}
library(readr)
clinical <- read_tsv("tcga_clinical_CLEANED.tsv")
```
If you got an error `could not find function "read_tsv"` at the previous step, it will because you don't have the `readr` package loaded. The RStudio output may indicate some problems with the data, but we should be able to proceed.
<div class="information">
If you get really stuck reading data into R, you can use the Import Dataset option from the File menu which will allow you to choose the parameters to read the data interactively
</div>
The Environment panel of RStudio (top-right) should show that an object called `clinical` has been created. This means that we can start doing analysis this dataset. The choice to call the object `clinical` was ours. We could have used any name instead of `clinical` but we chose something vaguely informative and memorable.
The dimensions of the object should should `7706 obs. of 420 variables`. This means the object we have created contains 7706 rows and 420 columns. Each row is a different observation; in this case a different biological sample. Each column records the value of a different variable. In R's terminology, we have just created a `tibble` which is a special case of something called a `data.frame`. As we will see the the object can contain either numbers or text in each column.
RStudio has a browsing functionality available that we can activate using the `View` function
```{r}
View(clinical)
```
Observations that are missing are indicated by an `NA`. It may look at first glance that the table mostly contains missing data. This is because some columns are only recorded for particular tumour types and not all tumour types recorded the same information.
# Data Exploration
We will now start to explore the data with a couple of different questions in mind:-
- **What are the differences in male/female split between different tumour types?**
- **Is there a difference in diagnosis age in different tumour types?**
The tasks required to do this will be greatly helped by the `dplyr` package.
```{r}
library(dplyr)
```
## Choosing what columns to analyse
The `select` function allows us to narrow down the number of variables we are interested in from 420. The first argument is always the name of the data frame. There are numerous different ways of specifying which column(s) you want, including listing the names of the columns of interest. Let's assume we already know the names of columns containing tumour type and gender.
```{r}
##Note that the spelling of "tumor" has to exactly match that found in the data
select(clinical,
tumor_tissue_site,
gender
)
```
We can reasonably guess that the Age information would be contained in a column that has "age" somewhere in the name. However, the column is not just called "age" or "Age"
```{r eval=FALSE}
select(clinical, age)
select(clinical, Age)
```
Without manually going through the columns, there are a few "helper" functions that we can employ
```{r}
select(clinical, starts_with("age"))
select(clinical, contains("age"))
select(clinical, contains("age_"))
```
Up to now we have not changed the underlying dataset. `select` is showing what the dataset looks like with the specified subset. If we want to make permanent changes we can create a variable
```{r}
analysis_data <- select(clinical,
tumor_tissue_site,
gender,
age_at_diagnosis)
```
## Restricting rows
The `select` function only performs the very specific task of letting you choose what columns you want to analyse. After using `select`, our dataset `analysis_data` still has all `r nrow(analysis_data)` rows.
The function to choose or restrict to the rows we might be interested in is called `filter`. We have to write a short R command to choose the rows.
e.g. we want only the male samples. Notice that two "=" signs are required. If you try and use the function with a single "=" R will print a helpful hint.
```{r}
filter(analysis_data, gender == "MALE")
```
and the females
```{r}
filter(analysis_data, gender == "FEMALE")
```
An equivalent, but somewhat unnecessary for this example, statement would be to ask for rows where the gender is **not equal** to `MALE`
```{r}
filter(analysis_data, gender != "MALE")
```
We can also restrict the data based on numeric columns by using `>`, `<` etc.
```{r}
filter(analysis_data, age_at_diagnosis > 80)
```
But what if we want males with Brain tumours? `dplyr` allows us to combine more than one condition if we separate them with a `,`. In computing this is known as an "and" statement and only rows where **both** statements are two will be shown. The line could be extended to include more than two tests if we wanted to.
```{r}
filter(analysis_data, gender == "MALE",tumor_tissue_site == "Brain")
```
Brain or Lung tumours? Using a `|` symbol instead of a `,` allows for *either* of two (or more) conditions to be `TRUE`.
```{r}
filter(analysis_data, tumor_tissue_site == "Brain" | tumor_tissue_site == "Lung")
```
To answer the question of how many males / females have a certain tumour type we could now use R statements such as:-
```{r}
filter(analysis_data, gender == "MALE",tumor_tissue_site == "Brain")
filter(analysis_data, gender == "FEMALE",tumor_tissue_site == "Brain")
```
and make a note of the number of observations included in the resulting data frame.
Although useful for data exploration, it would clearly be inefficient to get gender/tumour type counts in this way as we would have to repeat for all combinations of tumour type and gender. The function `count` can now give us exactly what we want.
```{r}
count(analysis_data,
tumor_tissue_site,
gender)
```
The `count` function is useful for tabulating the number of observations, but for other summary statistics a more general `sumamrise` function can be used. This can be used in conjunction with the basic summary statistics supported by base R. A summary statistic being something that can be applied to a series of numbers and produce a single number as a result. e.g. the average, minimum, maximum etc.
```{r}
summarise(analysis_data,
Average = mean(age_at_diagnosis),
min = min(age_at_diagnosis),
max = max(age_at_diagnosis))
```
However, we obviously have a problem due to missing values. If R sees and missing values in a column it will report the mean, minimum or maximum of that column as a missing value. Although this default behaviour can be changes, before proceeding we could also choose to remove any missing observations from the data. These are represented by a `NA` value, which is a special value and not a character label.
```{r}
filter(analysis_data, is.na(age_at_diagnosis) | is.na(tumor_tissue_site))
```
We want the opposite of the above; where the age of diagnosis and tumour site is **not** missing.
```{r}
analysis_data <- filter(analysis_data, !is.na(age_at_diagnosis), !is.na(tumor_tissue_site))
```
The summary will now work as expected.
```{r}
summarise(analysis_data,
Average = mean(age_at_diagnosis),
min = min(age_at_diagnosis),
max = max(age_at_diagnosis))
```
This might not be what we want in all circumstances, as the statistics can also be calculated on a per-tumour site basis.
```{r}
data_grouped <- group_by(analysis_data, tumor_tissue_site)
summarise(data_grouped,Average = mean(age_at_diagnosis),
min = min(age_at_diagnosis),
max = max(age_at_diagnosis) )
```
Further investigation of the data could also involve finding the observations with the maximum or minimum diagnosis age.
```{r}
## desc specifies descending order
arrange(analysis_data, desc(age_at_diagnosis))
```
```{r}
arrange(analysis_data, tumor_tissue_site, desc(age_at_diagnosis))
## could also add another sorting variable of gender if we wanted!
```
So far we have used several operations in isolation. However, the real joy (?) of `dplyr` is how different operations can be chained together. Lets say we just wanted female tumours.
```{r}
filter(analysis_data, gender == "FEMALE")
```
Our next step could be to remove the `gender` column since it is somewhat redundant.
```{r}
analysis_data2 <- filter(analysis_data, gender == "FEMALE")
select(analysis_data2, tumor_tissue_site, age_at_diagnosis)
## or
## select(analysis_data2, -gender)
```
The code would quickly get cumbersome if we wanted to include additional steps such as removing `NA` values. An alternative approach called "piping" is recommended.
```{r}
filter(analysis_data, gender == "FEMALE") %>% ## and then...
select(tumor_tissue_site, age_at_diagnosis) ## %>% and then...
```
## Overview of plotting
Our recommending way of creating plots in RStudio is to use the `ggplot2` package
```{r}
library(ggplot2)
```
A couple of useful references are given here-
- [ggplot2 reference guide](https://posit.co/wp-content/uploads/2022/10/data-visualization-1.pdf)
- [Flowchart for deciding on what graph type to use](https://www.data-to-viz.com/)
The general principle of creating a plot is the same regardless of what kind of plot we want to make
- specify the `data frame` containing the data we want to plot
- specify which columns in that data frame we want to use for various aesthetic aspects of the plot
- define the type of plot we want
- apply any additional format changes
In order to compare the age distributions of different tumour types we can imagine this being displayed as a boxplot with
- the age variable on the y-axis
- the type of tumour on the x-axis
this can be translated into `ggplot2` language as follows -
```{r}
ggplot(analysis_data, aes(x = tumor_tissue_site, y = age_at_diagnosis)) + geom_boxplot()
```
```{r}
ggplot(analysis_data, aes(x = tumor_tissue_site, y = age_at_diagnosis)) + geom_boxplot() + geom_jitter(width=0.1)
```
A bar plot would be a natural choice for showing the counts of male / female samples. The `geom_bar` plot will automatically count how many
```{r}
ggplot(analysis_data, aes(x = gender)) + geom_bar()
```
```{r}
bladder_data <- filter(analysis_data, tumor_tissue_site == "Bladder")
ggplot(bladder_data, aes(x = gender)) + geom_bar()
```
```{r}
ggplot(analysis_data, aes(x = gender)) + geom_bar() + facet_wrap(~tumor_tissue_site)
```