Adding UMI-tools #49
Comments
|
I think this would be a great addition 👍🏻 If you have any details (kits used for example), that would be helpful. |
|
QIAGEN kit details: https://resources.qiagenbioinformatics.com/manuals/biomedicalgenomicsanalysis/current/index.php?manual=Create_UMI_Reads_miRNA.html Regarding our slightly weird sequencing setup at the NGI:
|
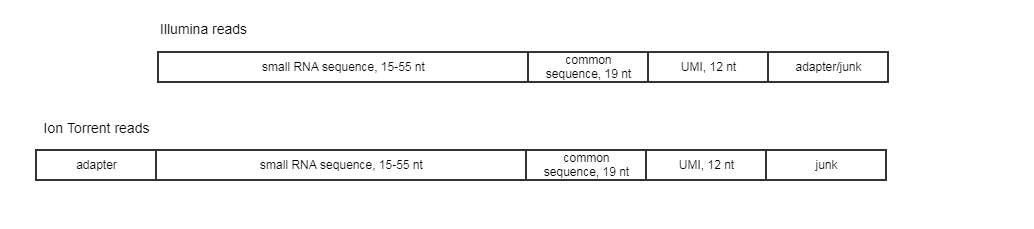
|
Hi, I am using that QIAGEN kit you found (btw. for few people would be handy that that common sequence is qiagen adapter sequence). |
|
@pfeiferl to be clear for future readers of the thread, that is the 3' adapter sequence as described in the Qiagen miRNAseq manual on page 53 (07/2020 version) and specified on these lines in the pipeline. Please correct me if I'm wrong. I'm currently working with a client who did 75bp sequencing so that the UMIs are in the sequence reads. I was going to try using UMI tools but haven't looked into it in depth. I'm a bit confused on how you specify the regex for extracting. Are you using the regular expression mode? Can you share your code for how you did the extract and dedup? Thank you |
|
@pcantalupo you are correct And umi extract: Yes, I am using regex - firstly the raw fastq files must NOT have trimmed adapter, you are searching for umi after it. umi_tools extract --stdin=in.fastq.gz --stdout=out.fastq.gz --extract-method=regex --bc-pattern='.+AACTGTAGGCACCATCAAT{s<=2}(? P<umi_1>.{12})(?P<discard_2>.*)'
Dedup just simply umi_tools dedup --method=unique -I in.bam -S out.bam |
|
Note that the main The rnaseq pipeline has just moved to DSL2, meaning that this functionality has been ported to Once the DSL2 stuff settles down we will want to start migrating all pipelines to DSL2. When we come to do this pipeline that'll mean that we can reuse the same modules to also run UMI tools in this pipeline. If we don't want to wait that long, we could always copy over code from the above ☝🏻 (but if we're not in a rush then I think it'd be better to wait). |
|
I have looked into that. Meanwhile extracting seems compatible with the pipeline and dedup BAM files can be used for some tools, there are some parts that will need some custom dedup before aligning. If somebody have time for this, I can help on the guiding how to implementing. I don't see a lot of time for doing it by myself in the next month, but who knows! |
Integrate the umi tools module already existing in nf-core into the smrnaseq pipeline. See Issue nf-core#49
|
@lpantano I'm currently looking into this. The UMI extract part is pretty straightforward to implement, but what steps do you refer to when you talk about custom deduplicating before aligning? Is a deduce step for the mapped hairpin and mature bam files (+ possibly for the genome alignment) not sufficient? |
|
Hi, dedup on bamfiles is not going to help some tools. If dedups happen in the fastq files, then it is fine. The tools targeted to a better quantification on miRNA will do a 'collapsing' step at the fastq level, where each sequence that is the same will be reported once in the output file (normally a fasta file), having the times where that sequence appeared in the read name. mirdeep, mirtop, mirtrace will do this. Any of them are working at the bam file levels in the same way than rnaseq. So botton line, if this could be done at fastq level then is fine, but if not, I don't think it will be that useful. The quantification from the bam file shouldn't be used for anything than just statistics about how many reads map to mature, hairpin or genome. Happy to set up a call to talk more, it is a little confusing, just because the history of smrnaseq analysis. thanks! |
|
So adding an additional step in genome mode that maps all reads, dedups the bam and converts it back to fastq would be an option? |
|
Assuming that UMI sequences are carried into aligned BAM files in the read headers, it should be fine to do alignment+UMI based deduplication, no? I don't really see why it has to be raw sequence based only? |
|
I think #164 adds exactly that now - so everyone could take a look at the feature and test it for inclusion 👍🏻 |
|
#303 adds UMI handling, please test this thourhgly! |
Hi all!
Firstly I would like to thank you for this awesome work. Secondly I have a request:
Please is it possible to add UMI-tools to environment (docker) to allow work with umi's?
Thank you for answer
The text was updated successfully, but these errors were encountered: