From d6c881660b6309e7c62055f568722cc8f488ba02 Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Thu, 22 Jul 2021 21:37:27 +0300
Subject: [PATCH 01/42] Adds workdir cleanup option (#238)
* Adds --cleanup option to clean work directory
* Adds explanatory comment to config for cleanup option
* Adds "Workdir cleanup" param to pipeline summary
* Adds --cleanup option description to help message
* Adds --cleanup option description to usage.md
* Adds singularity check in ci.yml (#240)
* Adds local singularity profile (#239)
* Adds profile to run with singularity locally
* Adds docks on how to run the pipeline locally
* Adds link to new docs to README.md
* Renames profile to singularity
* Adds singularity check in ci.yml (#240)
* Renames singularity_local -> singularity everywhere
Co-authored-by: cgpu <38183826+cgpu@users.noreply.github.com>
* Adds workflow.onComplete notifications
* Reduces ci tests (#241)
* Reduces ci tests to only run on pull_request, not push
* Reduces ci tests to only run with max_retries: 1
* Updates ci test nextflow version 19.04.0 -> 20.01.0
* Removes max_retries matrix option from ci tests
Co-authored-by: cgpu <38183826+cgpu@users.noreply.github.com>
---
.github/workflows/ci.yml | 28 +++++++++---
README.md | 1 +
conf/executors/singularity.config | 17 +++++++
docs/run_locally.md | 73 +++++++++++++++++++++++++++++++
docs/usage.md | 11 +++++
main.nf | 32 ++++++++++++++
nextflow.config | 14 +++++-
7 files changed, 169 insertions(+), 7 deletions(-)
create mode 100644 conf/executors/singularity.config
create mode 100644 docs/run_locally.md
diff --git a/.github/workflows/ci.yml b/.github/workflows/ci.yml
index ec42cf7e..4f670fae 100755
--- a/.github/workflows/ci.yml
+++ b/.github/workflows/ci.yml
@@ -1,14 +1,13 @@
name: splicing-pipelines-nf CI
# This workflow is triggered on pushes and PRs to the repository.
-on: [push, pull_request]
+on: [pull_request]
jobs:
- test:
+ docker:
runs-on: ubuntu-latest
strategy:
matrix:
- nxf_ver: ['19.04.0', '']
- max_retries: [1, 10]
+ nxf_ver: ['20.01.0', '']
steps:
- uses: actions/checkout@v1
- name: Install Nextflow
@@ -18,4 +17,23 @@ jobs:
sudo mv nextflow /usr/local/bin/
- name: Basic workflow tests
run: |
- nextflow run ${GITHUB_WORKSPACE} --max_retries ${{ matrix.max_retries }} -profile base,ultra_quick_test,docker
+ nextflow run ${GITHUB_WORKSPACE} -profile base,ultra_quick_test,docker
+ singularity:
+ runs-on: ubuntu-latest
+ strategy:
+ matrix:
+ singularity_version: ['3.6.4']
+ nxf_ver: ['20.01.0', '']
+ steps:
+ - uses: actions/checkout@v1
+ - uses: eWaterCycle/setup-singularity@v6
+ with:
+ singularity-version: ${{ matrix.singularity_version }}
+ - name: Install Nextflow
+ run: |
+ export NXF_VER=${{ matrix.nxf_ver }}
+ wget -qO- get.nextflow.io | bash
+ sudo mv nextflow /usr/local/bin/
+ - name: Basic workflow tests
+ run: |
+ nextflow run ${GITHUB_WORKSPACE} -profile base,ultra_quick_test,singularity
diff --git a/README.md b/README.md
index 85fa8e63..5bea0911 100755
--- a/README.md
+++ b/README.md
@@ -76,6 +76,7 @@ Documentation about the pipeline, found in the [`docs/`](docs) directory:
3. [Running the pipeline](docs/usage.md)
* [Running on Sumner](docs/run_on_sumner.md)
* [Running on CloudOS](docs/run_on_cloudos.md)
+ * [Running locally](docs/run_locally.md)
## Pipeline DAG
 diff --git a/conf/executors/singularity.config b/conf/executors/singularity.config
new file mode 100644
index 00000000..29db9760
--- /dev/null
+++ b/conf/executors/singularity.config
@@ -0,0 +1,17 @@
+/*
+ * -------------------------------------------------
+ * Nextflow config file for running pipeline with Singularity locally
+ * -------------------------------------------------
+ * Base config needed for running with -profile singularity
+ */
+
+params {
+ singularity_cache = "local_singularity_cache"
+}
+
+singularity {
+ enabled = true
+ cacheDir = params.singularity_cache
+ autoMounts = true
+}
+
diff --git a/docs/run_locally.md b/docs/run_locally.md
new file mode 100644
index 00000000..f630281f
--- /dev/null
+++ b/docs/run_locally.md
@@ -0,0 +1,73 @@
+# Run locally
+
+This guide provides information needed to run the pipeline in a local linux environment.
+
+## 0) Install Nextflow, Docker and/or Singularity
+
+### 0.1) Install Nextflow
+```bash
+wget -qO- https://get.nextflow.io | bash && sudo mv nextflow /usr/bin/
+```
+
+### 0.1) Install Docker
+See https://docs.docker.com/engine/install/ubuntu/
+
+
+### 0.2) Install Singularity
+See https://sylabs.io/guides/3.8/user-guide/quick_start.html and https://sylabs.io/guides/3.0/user-guide/installation.html
+
+Commands to install on CentOS:
+```bash
+sudo yum update -y && \
+sudo yum groupinstall -y 'Development Tools' && \
+sudo yum install -y \
+ openssl-devel \
+ libuuid-devel \
+ libseccomp-devel \
+ wget \
+ squashfs-tools
+
+export VERSION=1.16.6 OS=linux ARCH=amd64 && # adjust this as necessary \
+ wget https://dl.google.com/go/go$VERSION.$OS-$ARCH.tar.gz && \
+ sudo tar -C /usr/local -xzvf go$VERSION.$OS-$ARCH.tar.gz && \
+ rm go$VERSION.$OS-$ARCH.tar.gz
+
+echo 'export GOPATH=${HOME}/go' >> ~/.bashrc && \
+ echo 'export PATH=/usr/local/go/bin:${PATH}:${GOPATH}/bin' >> ~/.bashrc && \
+ source ~/.bashrc
+
+go get -u github.com/golang/dep/cmd/dep
+
+
+export VERSION=3.8.0 && # adjust this as necessary \
+ wget https://github.com/sylabs/singularity/releases/download/v${VERSION}/singularity-ce-${VERSION}.tar.gz && \
+ tar -xzf singularity-ce-${VERSION}.tar.gz && \
+ cd singularity-ce-${VERSION}
+
+./mconfig && \
+ make -C builddir && \
+ sudo make -C builddir install
+
+singularity help
+
+```
+
+## 1) Clone the git repository to local machine
+
+```bash
+git clone https://github.com/TheJacksonLaboratory/splicing-pipelines-nf
+cd splicing-pipelines-nf
+
+```
+
+## 2) Run the pipeline
+
+### 2.1) Quick test with Docker container engine
+```bash
+nextflow run . -profile ultra_quick_test,docker --cleanup
+```
+
+### 2.2) Quick test with Docker container engine
+```bash
+nextflow run . -profile ultra_quick_test,singularity --cleanup
+```
\ No newline at end of file
diff --git a/docs/usage.md b/docs/usage.md
index 7567e36a..862643c4 100755
--- a/docs/usage.md
+++ b/docs/usage.md
@@ -6,6 +6,9 @@ See [here](run_on_sumner.md)
## For running the pipeline on CloudOS
See [here](run_on_cloudos.md)
+## For running the pipeline locally
+See [here](run_locally.md)
+
# Nextflow parameters
Nextflow parameters can be provided in one of two ways:
@@ -184,6 +187,14 @@ Other:
--debug This option will enable echo of script execution into STDOUT with some additional
resource information (such as machine type, memory, cpu and disk space)
(default: false)
+ --cleanup This option will enable nextflow work folder cleanup upon pipeline successfull completion.
+ All intermediate files from nexftlow processes' workdirs will be cleared, staging folder with staged
+ files will not be cleared.
+ If pipeline is completed with errors or interrupted cleanup will not be executed. Following successfull run
+ resumed from the failed run with --cleanup option enabled will only clear folders of processess created in
+ the latest run, it will not clear cached folders coming from previous pipleine runs.
+ (default: false)
+
```
diff --git a/main.nf b/main.nf
index 75559072..d50d867f 100755
--- a/main.nf
+++ b/main.nf
@@ -116,6 +116,14 @@ def helpMessage() {
--debug This option will enable echo of script execution into STDOUT with some additional
resource information (such as machine type, memory, cpu and disk space)
(default: false)
+ --cleanup This option will enable nextflow work folder cleanup upon pipeline successfull completion.
+ All intermediate files from nexftlow processes' workdirs will be cleared, staging folder with staged
+ files will not be cleared.
+ If pipeline is completed with errors or interrupted cleanup will not be executed. Following successfull run
+ resumed from the failed run with --cleanup option enabled will only clear folders of processess created in
+ the latest run, it will not clear cached folders coming from previous pipleine runs.
+ (default: false)
+
See here for more info: https://github.com/TheJacksonLaboratory/splicing-pipelines-nf/blob/master/docs/usage.md
""".stripIndent()
@@ -199,6 +207,7 @@ log.info "Max time : ${params.max_time}"
log.info "Mega time : ${params.mega_time}"
log.info "Google Cloud disk-space : ${params.gc_disk_size}"
log.info "Debug : ${params.debug}"
+log.info "Workdir cleanup : ${params.cleanup}"
log.info ""
log.info "\n"
@@ -966,3 +975,26 @@ process collect_tool_versions_env2 {
def download_from(db) {
download_from.toLowerCase().contains(db)
}
+
+
+// Completion notification
+
+workflow.onComplete {
+
+ c_green = "\033[0;32m";
+ c_purple = "\033[0;35m";
+ c_red = "\033[0;31m";
+ c_reset = "\033[0m";
+
+ if (workflow.success) {
+ log.info "-${c_purple}[splicing-pipelines-nf]${c_green} Pipeline completed successfully${c_reset}-"
+ if (params.cleanup) {
+ log.info "-${c_purple}[splicing-pipelines-nf]${c_green} Cleanup: Working directory cleared from intermediate files generated with current run: '${workflow.workDir}' ${c_reset}-"
+ }
+ } else { // To be shown requires errorStrategy = 'finish'
+ log.info "-${c_purple}[splicing-pipelines-nf]${c_red} Pipeline completed with errors${c_reset}-"
+ if (params.cleanup) {
+ log.info "-${c_purple}[splicing-pipelines-nf]${c_red} Cleanup: Working directory was not cleared from intermediate files due to pipeline errors. You can re-use them with -resume option. ${c_reset}-"
+ }
+ }
+}
\ No newline at end of file
diff --git a/nextflow.config b/nextflow.config
index 68f62580..f858a6e3 100755
--- a/nextflow.config
+++ b/nextflow.config
@@ -51,6 +51,7 @@ params {
help = false
mega_time = 20.h
tracedir = "${params.outdir}/pipeline_info"
+ cleanup = false // if true will delete all intermediate files in work folder on workflow completion (not including staged files)
// Max resources
max_memory = 760.GB
@@ -64,7 +65,10 @@ params {
gls_boot_disk_size = 50.GB
}
+cleanup = params.cleanup
+
process {
+ errorStrategy = 'finish'
container = 'gcr.io/nextflow-250616/splicing-pipelines-nf:gawk'
withName: 'get_accession' {
container = 'lifebitai/download_reads:latest'
@@ -96,12 +100,18 @@ profiles {
standard {
includeConfig 'conf/executors/google.config'
}
- docker { docker.enabled = true }
+ docker {
+ docker.enabled = true
+ docker.runOptions = '-u $(id -u):$(id -g)' // to prevent files in workdir owned by root user
+ }
base { includeConfig 'conf/executors/base.config' }
sumner {
includeConfig 'conf/executors/base.config'
includeConfig 'conf/executors/sumner.config'
}
+ singularity {
+ includeConfig 'conf/executors/singularity.config'
+ }
MYC_MCF10A_0h_vs_MYC_MCF10A_8h { includeConfig 'conf/examples/MYC_MCF10A_0h_vs_MYC_MCF10A_8h.config' }
ultra_quick_test { includeConfig 'conf/examples/ultra_quick_test.config' }
}
@@ -122,4 +132,4 @@ trace {
enabled = true
file = "${params.tracedir}/execution_trace.txt"
fields = 'process,tag,name,status,exit,script'
-}
\ No newline at end of file
+}
From 70fab081899d6d1e544ebbb72fa2bbafda5fc66f Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Fri, 23 Jul 2021 19:19:22 +0300
Subject: [PATCH 02/42] Adds trimmomatic logs to multiqc report (#244)
* Adds trimmomatic logs to multiqc report
* Puts Trimmomatic above STAR in MultiQC report
---
examples/assets/multiqc_config.yaml | 6 +++++-
main.nf | 4 +++-
2 files changed, 8 insertions(+), 2 deletions(-)
diff --git a/examples/assets/multiqc_config.yaml b/examples/assets/multiqc_config.yaml
index 83884710..20b3b834 100755
--- a/examples/assets/multiqc_config.yaml
+++ b/examples/assets/multiqc_config.yaml
@@ -11,4 +11,8 @@ module_order:
target: ''
path_filters:
- '*_trimmed*_fastqc.zip'
- - star
\ No newline at end of file
+ - trimmomatic:
+ name: 'Trimmomatic'
+ path_filters:
+ - '*_trimmomatic.log'
+ - star
diff --git a/main.nf b/main.nf
index d50d867f..b2e32c7c 100755
--- a/main.nf
+++ b/main.nf
@@ -917,14 +917,16 @@ if (!params.bams) {
file (fastqc:'fastqc/*') from fastqc_results_trimmed.collect().ifEmpty([])
file ('alignment/*') from alignment_logs.collect().ifEmpty([])
file (multiqc_config) from multiqc_config
+ file ('trimmomatic/*') from trimmomatic_logs.collect()
output:
file "*multiqc_report.html" into multiqc_report
file "*_data/*"
+ file ('trimmomatic')
script:
"""
- multiqc . --config $multiqc_config -m fastqc -m star
+ multiqc . --config $multiqc_config -m fastqc -m star -m trimmomatic
cp multiqc_report.html ${run_prefix}_multiqc_report.html
"""
}
From 3c8bcdd0b7645f68b7fa6887fabd785bdaf19fe6 Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Wed, 11 Aug 2021 18:23:53 +0300
Subject: [PATCH 03/42] Updates all tools and moves docker containers to
anczukowlab (#248)
---
conf/examples/ultra_quick_test.config | 2 +-
containers/download_reads/environment.yml | 10 +++++-----
containers/fasp/Dockerfile | 2 +-
containers/splicing-pipelines-nf/Dockerfile | 13 ++++++++-----
.../splicing-pipelines-nf/environment.yml | 16 ++++++++--------
nextflow.config | 18 +++++++++---------
6 files changed, 32 insertions(+), 29 deletions(-)
diff --git a/conf/examples/ultra_quick_test.config b/conf/examples/ultra_quick_test.config
index 987cf5d9..a80c22c8 100755
--- a/conf/examples/ultra_quick_test.config
+++ b/conf/examples/ultra_quick_test.config
@@ -15,7 +15,7 @@ params {
// Genome references
gtf = 'https://lifebit-featured-datasets.s3-eu-west-1.amazonaws.com/projects/jax/splicing-pipelines-nf/genes.gtf'
- star_index = 'https://lifebit-featured-datasets.s3-eu-west-1.amazonaws.com/projects/jax/splicing-pipelines-nf/star.tar.gz'
+ star_index = 'https://lifebit-featured-datasets.s3-eu-west-1.amazonaws.com/projects/jax/splicing-pipelines-nf/star_2.7.9a_yeast_chr_I.tar.gz'
// Other
test = true
diff --git a/containers/download_reads/environment.yml b/containers/download_reads/environment.yml
index a85aad1f..9fa7f2af 100755
--- a/containers/download_reads/environment.yml
+++ b/containers/download_reads/environment.yml
@@ -5,8 +5,8 @@ channels:
- defaults
- anaconda
dependencies:
- - sra-tools=2.10.8
- - pigz=2.3.4
- - gdc-client=1.5.0
- - samtools=1.10
- - bedtools=2.29.2
\ No newline at end of file
+ - sra-tools=2.11.0
+ - pigz=2.6.0
+ - gdc-client=1.6.1
+ - samtools=1.13
+ - bedtools=2.30.0
\ No newline at end of file
diff --git a/containers/fasp/Dockerfile b/containers/fasp/Dockerfile
index 55694785..81f2d27f 100644
--- a/containers/fasp/Dockerfile
+++ b/containers/fasp/Dockerfile
@@ -17,7 +17,7 @@ RUN apt-get update \
&& cd fasp-scripts \
&& python setup.py install \
&& chmod +x fasp/scripts/* \
- && conda install samtools=1.11 -c bioconda -c conda-forge \
+ && conda install samtools=1.13 -c bioconda -c conda-forge \
&& conda clean -a
ENV PATH /fasp-scripts/fasp/scripts:$PATH
diff --git a/containers/splicing-pipelines-nf/Dockerfile b/containers/splicing-pipelines-nf/Dockerfile
index 5cfe4516..d479971a 100755
--- a/containers/splicing-pipelines-nf/Dockerfile
+++ b/containers/splicing-pipelines-nf/Dockerfile
@@ -9,12 +9,15 @@ ENV PATH /opt/conda/envs/splicing-pipelines-nf/bin:$PATH
COPY ./tagXSstrandedData.awk /usr/local/bin/
RUN chmod +x /usr/local/bin/tagXSstrandedData.awk
-# Install the latest stringtie & gffcompare
-RUN wget http://ccb.jhu.edu/software/stringtie/dl/stringtie-2.1.3b.Linux_x86_64.tar.gz -O stringtie.tar.gz && \
- tar xvzf stringtie.tar.gz && mv stringtie-2.1.3b.Linux_x86_64 stringtie && \
+# Install the latest stringtie, gffread & gffcompare
+RUN wget http://ccb.jhu.edu/software/stringtie/dl/stringtie-2.1.7.Linux_x86_64.tar.gz -O stringtie.tar.gz && \
+ tar xvzf stringtie.tar.gz && mv stringtie-2.1.7.Linux_x86_64 stringtie && \
rm stringtie.tar.gz && mv stringtie/prepDE.py stringtie/stringtie /usr/local/bin && \
- wget http://ccb.jhu.edu/software/stringtie/dl/gffcompare-0.11.6.Linux_x86_64.tar.gz -O gffcompare.tar.gz && \
- tar xvzf gffcompare.tar.gz && mv gffcompare-0.11.6.Linux_x86_64 gffcompare && \
+ wget http://ccb.jhu.edu/software/stringtie/dl/gffread-0.12.7.Linux_x86_64.tar.gz -O gffread.tar.gz && \
+ tar xvzf gffread.tar.gz && mv gffread-0.12.7.Linux_x86_64 gffread && \
+ rm gffread.tar.gz && mv gffread/gffread /usr/local/bin/ && \
+ wget http://ccb.jhu.edu/software/stringtie/dl/gffcompare-0.12.6.Linux_x86_64.tar.gz -O gffcompare.tar.gz && \
+ tar xvzf gffcompare.tar.gz && mv gffcompare-0.12.6.Linux_x86_64 gffcompare && \
rm gffcompare.tar.gz && mv gffcompare/gffcompare gffcompare/trmap /usr/local/bin/
# Install gawk to fix https://github.com/TheJacksonLaboratory/splicing-pipelines-nf/issues/120
RUN apt-get install -y gawk
\ No newline at end of file
diff --git a/containers/splicing-pipelines-nf/environment.yml b/containers/splicing-pipelines-nf/environment.yml
index 9e8b37f0..d306ebab 100755
--- a/containers/splicing-pipelines-nf/environment.yml
+++ b/containers/splicing-pipelines-nf/environment.yml
@@ -6,13 +6,13 @@ channels:
dependencies:
- fastqc=0.11.9
- trimmomatic=0.39
- - star=2.7.3
- - samtools=1.10
- - deeptools=3.4.0
- - multiqc=1.8
- - gffread=0.11.7
- - bioconductor-rtracklayer=1.46.0
+ - star=2.7.9a
+ - samtools=1.13
+ - deeptools=3.5.1
+ - multiqc=1.11
+ - bioconductor-rtracklayer=1.52.0
# Now installed via executable in `Dockerfile`
- # - stringtie=2.1.2
- # - gffcompare=0.11.2
\ No newline at end of file
+ # - stringtie=2.1.7
+ # - gffread=0.12.7 (version not available on conda)
+ # - gffcompare=0.12.6
\ No newline at end of file
diff --git a/nextflow.config b/nextflow.config
index f858a6e3..e23aec63 100755
--- a/nextflow.config
+++ b/nextflow.config
@@ -69,30 +69,30 @@ cleanup = params.cleanup
process {
errorStrategy = 'finish'
- container = 'gcr.io/nextflow-250616/splicing-pipelines-nf:gawk'
+ container = 'anczukowlab/splicing-pipelines-nf:3.0'
withName: 'get_accession' {
- container = 'lifebitai/download_reads:latest'
+ container = 'anczukowlab/download_reads:2.0'
}
withName: 'gen3_drs_fasp' {
- container = 'quay.io/lifebitai/lifebit-ai-fasp:latest'
+ container = 'anczukowlab/lifebit-ai-fasp:v1.1'
}
withName: 'get_tcga_bams' {
- container = 'lifebitai/download_reads:latest'
+ container = 'anczukowlab/download_reads:2.0'
}
withName: 'bamtofastq' {
- container = 'lifebitai/download_reads:latest'
+ container = 'anczukowlab/download_reads:2.0'
}
withName: 'rmats' {
- container = 'lifebitai/splicing-rmats:4.1.1'
+ container = 'anczukowlab/splicing-rmats:4.1.1'
}
withName: 'paired_rmats' {
- container = 'lifebitai/splicing-rmats:4.1.1'
+ container = 'anczukowlab/splicing-rmats:4.1.1'
}
withName: 'collect_tool_versions_env1' {
- container = 'gcr.io/nextflow-250616/splicing-pipelines-nf:gawk'
+ container = 'anczukowlab/splicing-pipelines-nf:3.0'
}
withName: 'collect_tool_versions_env2' {
- container = 'lifebitai/splicing-rmats:4.1.1'
+ container = 'anczukowlab/splicing-rmats:4.1.1'
}
}
From 3141ca177022b0399cf93f74f4e1bc3db9613d76 Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Fri, 13 Aug 2021 15:00:09 -0400
Subject: [PATCH 04/42] Update usage.md
Best solution to #246 is to update documentation.
---
docs/usage.md | 1 +
1 file changed, 1 insertion(+)
diff --git a/docs/usage.md b/docs/usage.md
index 862643c4..f330cd51 100755
--- a/docs/usage.md
+++ b/docs/usage.md
@@ -178,6 +178,7 @@ Other:
--skipMultiQC Skip MultiQC (bool)
(default: false)
--outdir The output directory where the results will be saved (string)
+ On Sumner, this must be set in the main.pbs. NF_splicing_pipeline.config will not overwrite main.pbs.
(default: directory where you submit the job)
--mega_time Sets time limit for processes withLabel 'mega_memory' in the main.nf using the base.config (time unit)
Make sure '#SBATCH -t' in 'main.pbs' is appropriately set if you are changing this parameter.
From 33e5d8aa1693be661a0074a101232510aeeca422 Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Fri, 13 Aug 2021 15:12:15 -0400
Subject: [PATCH 05/42] Update usage.md
---
docs/usage.md | 5 ++---
1 file changed, 2 insertions(+), 3 deletions(-)
diff --git a/docs/usage.md b/docs/usage.md
index f330cd51..fefdd545 100755
--- a/docs/usage.md
+++ b/docs/usage.md
@@ -117,6 +117,7 @@ Main arguments:
(default: false)
--stranded Specifies that the input is stranded ('first-strand', 'second-strand', false (aka unstranded))
(default: 'first-strand')
+ 'first-strand' refers to RF/fr-firststrand in this pipeline.
--readlength Read length - Note that all reads will be cropped to this length(int)
(default: no read length specified)
-profile Configuration profile to use. Can use multiple (comma separated, string)
@@ -239,7 +240,5 @@ Here `query_list.csv` should look something like -
```csv
file_name,sequencing_assay,data_format,file_name,sample_id,participant_id,tissue,age,gender
-GTEX-11EM3-1326-SM-5N9C6,RNA-Seq,bam,GTEX-11EM3-1326-SM-5N9C6.Aligned.sortedByCoord.out.patched.md.bam,GTEX-11EM3-1326-SM-5N9C6,GTEX-11EM3,Breast,21,Female
-GTEX-RU1J-0626-SM-4WAWY,RNA-Seq,bam,GTEX-RU1J-0626-SM-4WAWY.Aligned.sortedByCoord.out.patched.md.bam,GTEX-RU1J-0626-SM-4WAWY,GTEX-RU1J,Breast,21,Female
-GTEX-ZTPG-2826-SM-57WGA,RNA-Seq,bam,GTEX-ZTPG-2826-SM-57WGA.Aligned.sortedByCoord.out.patched.md.bam,GTEX-ZTPG-2826-SM-57WGA,GTEX-ZTPG,Breast,21,Female
+GTEX-PPPP-XXX-XX-XXXXX,RNA-Seq,bam,GTEX-PPPP-XXX-XX-XXXXX.Aligned.sortedByCoord.out.patched.md.bam,GTEX-PPPP-XXX-XX-XXXXX,GTEX-PPPP,Breast,21,Female
```
From 483e1f0d0e1810fefb2500837b9f26bc53d700d9 Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Fri, 13 Aug 2021 15:27:21 -0400
Subject: [PATCH 06/42] Update run_on_sumner.md
Adding some clarification to address #247
---
docs/run_on_sumner.md | 9 ++++++---
1 file changed, 6 insertions(+), 3 deletions(-)
diff --git a/docs/run_on_sumner.md b/docs/run_on_sumner.md
index a3c206a1..9c359211 100755
--- a/docs/run_on_sumner.md
+++ b/docs/run_on_sumner.md
@@ -19,6 +19,7 @@ This pipeline can be run on Sumner in three ways:
2. Input a 'reads' file to input fastq files and run pipeline until STAR mapping step with 'test' parameter.
3. Input a 'bams' file to input bams and run steps of pipeline following STAR mapping (Stringtie and rMATS).
+The `cacheDir` stores singularity images. This is set in splicing-pipelines-nf/conf/executors/sumner.config. For non-Anczukow users, this should be changed to a home directory.
## Running full pipeline with FASTQ input
### 1. Create a new run directory
@@ -81,14 +82,16 @@ If you already created a `NF_splicing_pipeline.config` during the trim test, you
- Each time you run the pipeline, go through all possible parameters to ensure you are creating a config ideal for your data. If you do not specify a value for a parameter, the default will be used. All parameters used can be found in the `log` file. WHEN IN DOUBT, SPECIFY ALL PARAMETERS
-- You must name your config file `NF_splicing_pipeline.config`
+- You must name your config file `NF_splicing_pipeline.config` (as specified in main.pbs)
- Your `NF_splicing_pipeline.config` must be in the directory that you are running your analysis.
-- The `readlength` here should be the length of the reads - if read leangth is not a multiple of 5 (ex- 76 or 151), set 'readlength' to nearest multiple of 5 (ex- 75 or 150). This extra base is an artifact of Illumina sequencing
+- The `readlength` here should be the length of the reads - if read length is not a multiple of 5 (ex- 76 or 151), set 'readlength' to nearest multiple of 5 (ex- 75 or 150). This extra base is an artifact of Illumina sequencing
- To run full pipeline, you **must** specify the following: `reads.csv`, `rmats_pairs.txt`, `readlength`, `assembly_name`, `star_index`, and `reference gtf`. This string can be a relative path from the directory in which you run Nextflow in, an absolute path or a link.
+- The star indexes must be generated prior to executing the pipeline (this is a separate step).
+
- Currently, the two options for genomes are hg38 and mm10. If you wish to use a newer version of the genome, you will need to add this to the post-processing script.
### 5. Run the pipeline!
@@ -106,7 +109,7 @@ Whereas parameters are set on the command-line using double dash options eg `--r
You can see some of these options [here](https://www.nextflow.io/docs/latest/tracing.html) in the Nextflow documentation.
-Some useful ones include:
+Some useful ones include (specified in main.pbs):
- `-resume` which will [resume](https://www.nextflow.io/docs/latest/getstarted.html?highlight=resume#modify-and-resume) any cached processes that have not been changed
- `-with-trace` eg `-with-trace trace.txt` which gives a [trace report](https://www.nextflow.io/docs/latest/tracing.html?highlight=dag#trace-report) for resource consumption by the pipeline
- `-with-dag` eg `-with-dag flowchart.png` which produces the [DAG visualisation](https://www.nextflow.io/docs/latest/tracing.html?highlight=dag#dag-visualisation) graph showing each of the different processes and the connections between them (the channels)
From 9027151f1c1ffef03afd3cd0f447c83d5859ad62 Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Fri, 13 Aug 2021 15:58:44 -0400
Subject: [PATCH 07/42] Update run_on_sumner.md
Adding the description to run pipeline with bams.csv to documentation
---
docs/run_on_sumner.md | 84 +++++++++++++++++++++++++++++++++++++++----
1 file changed, 78 insertions(+), 6 deletions(-)
diff --git a/docs/run_on_sumner.md b/docs/run_on_sumner.md
index 9c359211..91d6a9f5 100755
--- a/docs/run_on_sumner.md
+++ b/docs/run_on_sumner.md
@@ -15,9 +15,9 @@ git pull
Note: if you have not successfully completed the pipeline test, see [here](../README.md##quick-start-on-sumner-jaxs-hpc)
This pipeline can be run on Sumner in three ways:
- 1. Input a 'reads' file to input fastq files and run the pipeline in its entirety.
- 2. Input a 'reads' file to input fastq files and run pipeline until STAR mapping step with 'test' parameter.
- 3. Input a 'bams' file to input bams and run steps of pipeline following STAR mapping (Stringtie and rMATS).
+ 1. Input a `reads.csv` file to input fastq files and run the pipeline in its entirety.
+ 2. Input a `reads.csv` file to input fastq files and run pipeline until STAR mapping step with `--test` parameter set to `true`.
+ 3. Input a `bams.csv` file to input bams and run steps of pipeline following STAR mapping (Stringtie and rMATS).
The `cacheDir` stores singularity images. This is set in splicing-pipelines-nf/conf/executors/sumner.config. For non-Anczukow users, this should be changed to a home directory.
@@ -65,7 +65,7 @@ Each rMATS comparison must be specified with a comparison name as well as the `s
comparison2_id[space]sample3replicate1,sample3replicate2,sample3replicate3[space]sample4replicate1,sample4replicate1,sample4replicate1
```
- #### B1 only, no rMATS comparison (if this is run, set '--Statoff' parameter to 'true'):
+ #### B1 only, no rMATS comparison (if this is run, set '--statoff' parameter to 'true'):
```
comparison_id[space]sample1,sample2,sample3
```
@@ -76,11 +76,11 @@ Each rMATS comparison must be specified with a comparison name as well as the `s
This config file will be specific to your user and analysis. **You do not need to edit the pipeline code to configure the pipeline**. Descriptions of all possible parameters and their default values can be found [here](usage.md#all-available-parameters).
-If you already created a `NF_splicing_pipeline.config` during the trim test, you can modify it. Otherwise, to create your own custom config (to specify your input parameters) you can copy and edit this [example config](../conf/examples/MYC_MCF10A_0h_vs_MYC_MCF10A_8h.config) file.
+To create your own custom config (to specify your input parameters) you can copy and edit this [example config](../conf/examples/MYC_MCF10A_0h_vs_MYC_MCF10A_8h.config) file.
**VERY IMPORTANT NOTES***
-- Each time you run the pipeline, go through all possible parameters to ensure you are creating a config ideal for your data. If you do not specify a value for a parameter, the default will be used. All parameters used can be found in the `log` file. WHEN IN DOUBT, SPECIFY ALL PARAMETERS
+- Each time you run the pipeline, go through all possible parameters to ensure you are creating a config ideal for your data. If you do not specify a value for a parameter, the default will be used. All parameters used can be found in the `log` file. WHEN IN DOUBT, SPECIFY ALL PARAMETERS!
- You must name your config file `NF_splicing_pipeline.config` (as specified in main.pbs)
@@ -103,6 +103,78 @@ Run the pipeline!
sbatch /projects/anczukow-lab/splicing_pipeline/splicing-pipelines-nf/main.pbs
```
+## Running Stringtie and rMATS with BAM input
+### 1. Create a new run directory
+
+Create a new run directory within the appropriate dataset directory with the following format: runNumber_initials_date `run1_LU_20200519` (Example RUN Directory - `/projects/anczukow-lab/NGS_analysis/Dataset_4_MYC_MCF10A/run1_LU_20200519`).
+
+### 2. Create/Locate `bams.csv` file for your dataset
+
+Input reads are specified by the `bams` input parameter, specifying a path to a CSV file.
+
+- (create example) must contain columns for `sample_id`, `bam`, and `bam.bai`
+
+The 'bams.csv' column names must match the above example. The `sample_id` can be anything, however each must be unique. The `bam` column should contain the path to BAM files. The `bam.bai` column should contain the path to BAM.BAI files. You can create this on your local computer in excel and use WinSCP to move it to Sumner, or use create it using `nano` on the cluster.
+
+Supplying the `bams.csv` will signal to the pipeline to skip the first steps of the pipeline and start with Stringtie. No other parameter is needed.
+
+### 3. Create `rmats_pairs.txt` input file
+
+Each rMATS comparison must be specified with a comparison name as well as the `sample_id` as specified in the [`bams.csv`](create example) file. See example [`rmats_pairs.txt`](../examples/human_test/rmats_pairs.txt). Each line in the file corresponds to an rMATS execution. The first column corresponds to a unique name/id for the rMATS comparison (this will be used for the output folder/file names).
+
+* Replicates should be comma separated and the samples for the `b1` / `b2` files i.e. case and control should be space separated
+
diff --git a/conf/executors/singularity.config b/conf/executors/singularity.config
new file mode 100644
index 00000000..29db9760
--- /dev/null
+++ b/conf/executors/singularity.config
@@ -0,0 +1,17 @@
+/*
+ * -------------------------------------------------
+ * Nextflow config file for running pipeline with Singularity locally
+ * -------------------------------------------------
+ * Base config needed for running with -profile singularity
+ */
+
+params {
+ singularity_cache = "local_singularity_cache"
+}
+
+singularity {
+ enabled = true
+ cacheDir = params.singularity_cache
+ autoMounts = true
+}
+
diff --git a/docs/run_locally.md b/docs/run_locally.md
new file mode 100644
index 00000000..f630281f
--- /dev/null
+++ b/docs/run_locally.md
@@ -0,0 +1,73 @@
+# Run locally
+
+This guide provides information needed to run the pipeline in a local linux environment.
+
+## 0) Install Nextflow, Docker and/or Singularity
+
+### 0.1) Install Nextflow
+```bash
+wget -qO- https://get.nextflow.io | bash && sudo mv nextflow /usr/bin/
+```
+
+### 0.1) Install Docker
+See https://docs.docker.com/engine/install/ubuntu/
+
+
+### 0.2) Install Singularity
+See https://sylabs.io/guides/3.8/user-guide/quick_start.html and https://sylabs.io/guides/3.0/user-guide/installation.html
+
+Commands to install on CentOS:
+```bash
+sudo yum update -y && \
+sudo yum groupinstall -y 'Development Tools' && \
+sudo yum install -y \
+ openssl-devel \
+ libuuid-devel \
+ libseccomp-devel \
+ wget \
+ squashfs-tools
+
+export VERSION=1.16.6 OS=linux ARCH=amd64 && # adjust this as necessary \
+ wget https://dl.google.com/go/go$VERSION.$OS-$ARCH.tar.gz && \
+ sudo tar -C /usr/local -xzvf go$VERSION.$OS-$ARCH.tar.gz && \
+ rm go$VERSION.$OS-$ARCH.tar.gz
+
+echo 'export GOPATH=${HOME}/go' >> ~/.bashrc && \
+ echo 'export PATH=/usr/local/go/bin:${PATH}:${GOPATH}/bin' >> ~/.bashrc && \
+ source ~/.bashrc
+
+go get -u github.com/golang/dep/cmd/dep
+
+
+export VERSION=3.8.0 && # adjust this as necessary \
+ wget https://github.com/sylabs/singularity/releases/download/v${VERSION}/singularity-ce-${VERSION}.tar.gz && \
+ tar -xzf singularity-ce-${VERSION}.tar.gz && \
+ cd singularity-ce-${VERSION}
+
+./mconfig && \
+ make -C builddir && \
+ sudo make -C builddir install
+
+singularity help
+
+```
+
+## 1) Clone the git repository to local machine
+
+```bash
+git clone https://github.com/TheJacksonLaboratory/splicing-pipelines-nf
+cd splicing-pipelines-nf
+
+```
+
+## 2) Run the pipeline
+
+### 2.1) Quick test with Docker container engine
+```bash
+nextflow run . -profile ultra_quick_test,docker --cleanup
+```
+
+### 2.2) Quick test with Docker container engine
+```bash
+nextflow run . -profile ultra_quick_test,singularity --cleanup
+```
\ No newline at end of file
diff --git a/docs/usage.md b/docs/usage.md
index 7567e36a..862643c4 100755
--- a/docs/usage.md
+++ b/docs/usage.md
@@ -6,6 +6,9 @@ See [here](run_on_sumner.md)
## For running the pipeline on CloudOS
See [here](run_on_cloudos.md)
+## For running the pipeline locally
+See [here](run_locally.md)
+
# Nextflow parameters
Nextflow parameters can be provided in one of two ways:
@@ -184,6 +187,14 @@ Other:
--debug This option will enable echo of script execution into STDOUT with some additional
resource information (such as machine type, memory, cpu and disk space)
(default: false)
+ --cleanup This option will enable nextflow work folder cleanup upon pipeline successfull completion.
+ All intermediate files from nexftlow processes' workdirs will be cleared, staging folder with staged
+ files will not be cleared.
+ If pipeline is completed with errors or interrupted cleanup will not be executed. Following successfull run
+ resumed from the failed run with --cleanup option enabled will only clear folders of processess created in
+ the latest run, it will not clear cached folders coming from previous pipleine runs.
+ (default: false)
+
```
diff --git a/main.nf b/main.nf
index 75559072..d50d867f 100755
--- a/main.nf
+++ b/main.nf
@@ -116,6 +116,14 @@ def helpMessage() {
--debug This option will enable echo of script execution into STDOUT with some additional
resource information (such as machine type, memory, cpu and disk space)
(default: false)
+ --cleanup This option will enable nextflow work folder cleanup upon pipeline successfull completion.
+ All intermediate files from nexftlow processes' workdirs will be cleared, staging folder with staged
+ files will not be cleared.
+ If pipeline is completed with errors or interrupted cleanup will not be executed. Following successfull run
+ resumed from the failed run with --cleanup option enabled will only clear folders of processess created in
+ the latest run, it will not clear cached folders coming from previous pipleine runs.
+ (default: false)
+
See here for more info: https://github.com/TheJacksonLaboratory/splicing-pipelines-nf/blob/master/docs/usage.md
""".stripIndent()
@@ -199,6 +207,7 @@ log.info "Max time : ${params.max_time}"
log.info "Mega time : ${params.mega_time}"
log.info "Google Cloud disk-space : ${params.gc_disk_size}"
log.info "Debug : ${params.debug}"
+log.info "Workdir cleanup : ${params.cleanup}"
log.info ""
log.info "\n"
@@ -966,3 +975,26 @@ process collect_tool_versions_env2 {
def download_from(db) {
download_from.toLowerCase().contains(db)
}
+
+
+// Completion notification
+
+workflow.onComplete {
+
+ c_green = "\033[0;32m";
+ c_purple = "\033[0;35m";
+ c_red = "\033[0;31m";
+ c_reset = "\033[0m";
+
+ if (workflow.success) {
+ log.info "-${c_purple}[splicing-pipelines-nf]${c_green} Pipeline completed successfully${c_reset}-"
+ if (params.cleanup) {
+ log.info "-${c_purple}[splicing-pipelines-nf]${c_green} Cleanup: Working directory cleared from intermediate files generated with current run: '${workflow.workDir}' ${c_reset}-"
+ }
+ } else { // To be shown requires errorStrategy = 'finish'
+ log.info "-${c_purple}[splicing-pipelines-nf]${c_red} Pipeline completed with errors${c_reset}-"
+ if (params.cleanup) {
+ log.info "-${c_purple}[splicing-pipelines-nf]${c_red} Cleanup: Working directory was not cleared from intermediate files due to pipeline errors. You can re-use them with -resume option. ${c_reset}-"
+ }
+ }
+}
\ No newline at end of file
diff --git a/nextflow.config b/nextflow.config
index 68f62580..f858a6e3 100755
--- a/nextflow.config
+++ b/nextflow.config
@@ -51,6 +51,7 @@ params {
help = false
mega_time = 20.h
tracedir = "${params.outdir}/pipeline_info"
+ cleanup = false // if true will delete all intermediate files in work folder on workflow completion (not including staged files)
// Max resources
max_memory = 760.GB
@@ -64,7 +65,10 @@ params {
gls_boot_disk_size = 50.GB
}
+cleanup = params.cleanup
+
process {
+ errorStrategy = 'finish'
container = 'gcr.io/nextflow-250616/splicing-pipelines-nf:gawk'
withName: 'get_accession' {
container = 'lifebitai/download_reads:latest'
@@ -96,12 +100,18 @@ profiles {
standard {
includeConfig 'conf/executors/google.config'
}
- docker { docker.enabled = true }
+ docker {
+ docker.enabled = true
+ docker.runOptions = '-u $(id -u):$(id -g)' // to prevent files in workdir owned by root user
+ }
base { includeConfig 'conf/executors/base.config' }
sumner {
includeConfig 'conf/executors/base.config'
includeConfig 'conf/executors/sumner.config'
}
+ singularity {
+ includeConfig 'conf/executors/singularity.config'
+ }
MYC_MCF10A_0h_vs_MYC_MCF10A_8h { includeConfig 'conf/examples/MYC_MCF10A_0h_vs_MYC_MCF10A_8h.config' }
ultra_quick_test { includeConfig 'conf/examples/ultra_quick_test.config' }
}
@@ -122,4 +132,4 @@ trace {
enabled = true
file = "${params.tracedir}/execution_trace.txt"
fields = 'process,tag,name,status,exit,script'
-}
\ No newline at end of file
+}
From 70fab081899d6d1e544ebbb72fa2bbafda5fc66f Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Fri, 23 Jul 2021 19:19:22 +0300
Subject: [PATCH 02/42] Adds trimmomatic logs to multiqc report (#244)
* Adds trimmomatic logs to multiqc report
* Puts Trimmomatic above STAR in MultiQC report
---
examples/assets/multiqc_config.yaml | 6 +++++-
main.nf | 4 +++-
2 files changed, 8 insertions(+), 2 deletions(-)
diff --git a/examples/assets/multiqc_config.yaml b/examples/assets/multiqc_config.yaml
index 83884710..20b3b834 100755
--- a/examples/assets/multiqc_config.yaml
+++ b/examples/assets/multiqc_config.yaml
@@ -11,4 +11,8 @@ module_order:
target: ''
path_filters:
- '*_trimmed*_fastqc.zip'
- - star
\ No newline at end of file
+ - trimmomatic:
+ name: 'Trimmomatic'
+ path_filters:
+ - '*_trimmomatic.log'
+ - star
diff --git a/main.nf b/main.nf
index d50d867f..b2e32c7c 100755
--- a/main.nf
+++ b/main.nf
@@ -917,14 +917,16 @@ if (!params.bams) {
file (fastqc:'fastqc/*') from fastqc_results_trimmed.collect().ifEmpty([])
file ('alignment/*') from alignment_logs.collect().ifEmpty([])
file (multiqc_config) from multiqc_config
+ file ('trimmomatic/*') from trimmomatic_logs.collect()
output:
file "*multiqc_report.html" into multiqc_report
file "*_data/*"
+ file ('trimmomatic')
script:
"""
- multiqc . --config $multiqc_config -m fastqc -m star
+ multiqc . --config $multiqc_config -m fastqc -m star -m trimmomatic
cp multiqc_report.html ${run_prefix}_multiqc_report.html
"""
}
From 3c8bcdd0b7645f68b7fa6887fabd785bdaf19fe6 Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Wed, 11 Aug 2021 18:23:53 +0300
Subject: [PATCH 03/42] Updates all tools and moves docker containers to
anczukowlab (#248)
---
conf/examples/ultra_quick_test.config | 2 +-
containers/download_reads/environment.yml | 10 +++++-----
containers/fasp/Dockerfile | 2 +-
containers/splicing-pipelines-nf/Dockerfile | 13 ++++++++-----
.../splicing-pipelines-nf/environment.yml | 16 ++++++++--------
nextflow.config | 18 +++++++++---------
6 files changed, 32 insertions(+), 29 deletions(-)
diff --git a/conf/examples/ultra_quick_test.config b/conf/examples/ultra_quick_test.config
index 987cf5d9..a80c22c8 100755
--- a/conf/examples/ultra_quick_test.config
+++ b/conf/examples/ultra_quick_test.config
@@ -15,7 +15,7 @@ params {
// Genome references
gtf = 'https://lifebit-featured-datasets.s3-eu-west-1.amazonaws.com/projects/jax/splicing-pipelines-nf/genes.gtf'
- star_index = 'https://lifebit-featured-datasets.s3-eu-west-1.amazonaws.com/projects/jax/splicing-pipelines-nf/star.tar.gz'
+ star_index = 'https://lifebit-featured-datasets.s3-eu-west-1.amazonaws.com/projects/jax/splicing-pipelines-nf/star_2.7.9a_yeast_chr_I.tar.gz'
// Other
test = true
diff --git a/containers/download_reads/environment.yml b/containers/download_reads/environment.yml
index a85aad1f..9fa7f2af 100755
--- a/containers/download_reads/environment.yml
+++ b/containers/download_reads/environment.yml
@@ -5,8 +5,8 @@ channels:
- defaults
- anaconda
dependencies:
- - sra-tools=2.10.8
- - pigz=2.3.4
- - gdc-client=1.5.0
- - samtools=1.10
- - bedtools=2.29.2
\ No newline at end of file
+ - sra-tools=2.11.0
+ - pigz=2.6.0
+ - gdc-client=1.6.1
+ - samtools=1.13
+ - bedtools=2.30.0
\ No newline at end of file
diff --git a/containers/fasp/Dockerfile b/containers/fasp/Dockerfile
index 55694785..81f2d27f 100644
--- a/containers/fasp/Dockerfile
+++ b/containers/fasp/Dockerfile
@@ -17,7 +17,7 @@ RUN apt-get update \
&& cd fasp-scripts \
&& python setup.py install \
&& chmod +x fasp/scripts/* \
- && conda install samtools=1.11 -c bioconda -c conda-forge \
+ && conda install samtools=1.13 -c bioconda -c conda-forge \
&& conda clean -a
ENV PATH /fasp-scripts/fasp/scripts:$PATH
diff --git a/containers/splicing-pipelines-nf/Dockerfile b/containers/splicing-pipelines-nf/Dockerfile
index 5cfe4516..d479971a 100755
--- a/containers/splicing-pipelines-nf/Dockerfile
+++ b/containers/splicing-pipelines-nf/Dockerfile
@@ -9,12 +9,15 @@ ENV PATH /opt/conda/envs/splicing-pipelines-nf/bin:$PATH
COPY ./tagXSstrandedData.awk /usr/local/bin/
RUN chmod +x /usr/local/bin/tagXSstrandedData.awk
-# Install the latest stringtie & gffcompare
-RUN wget http://ccb.jhu.edu/software/stringtie/dl/stringtie-2.1.3b.Linux_x86_64.tar.gz -O stringtie.tar.gz && \
- tar xvzf stringtie.tar.gz && mv stringtie-2.1.3b.Linux_x86_64 stringtie && \
+# Install the latest stringtie, gffread & gffcompare
+RUN wget http://ccb.jhu.edu/software/stringtie/dl/stringtie-2.1.7.Linux_x86_64.tar.gz -O stringtie.tar.gz && \
+ tar xvzf stringtie.tar.gz && mv stringtie-2.1.7.Linux_x86_64 stringtie && \
rm stringtie.tar.gz && mv stringtie/prepDE.py stringtie/stringtie /usr/local/bin && \
- wget http://ccb.jhu.edu/software/stringtie/dl/gffcompare-0.11.6.Linux_x86_64.tar.gz -O gffcompare.tar.gz && \
- tar xvzf gffcompare.tar.gz && mv gffcompare-0.11.6.Linux_x86_64 gffcompare && \
+ wget http://ccb.jhu.edu/software/stringtie/dl/gffread-0.12.7.Linux_x86_64.tar.gz -O gffread.tar.gz && \
+ tar xvzf gffread.tar.gz && mv gffread-0.12.7.Linux_x86_64 gffread && \
+ rm gffread.tar.gz && mv gffread/gffread /usr/local/bin/ && \
+ wget http://ccb.jhu.edu/software/stringtie/dl/gffcompare-0.12.6.Linux_x86_64.tar.gz -O gffcompare.tar.gz && \
+ tar xvzf gffcompare.tar.gz && mv gffcompare-0.12.6.Linux_x86_64 gffcompare && \
rm gffcompare.tar.gz && mv gffcompare/gffcompare gffcompare/trmap /usr/local/bin/
# Install gawk to fix https://github.com/TheJacksonLaboratory/splicing-pipelines-nf/issues/120
RUN apt-get install -y gawk
\ No newline at end of file
diff --git a/containers/splicing-pipelines-nf/environment.yml b/containers/splicing-pipelines-nf/environment.yml
index 9e8b37f0..d306ebab 100755
--- a/containers/splicing-pipelines-nf/environment.yml
+++ b/containers/splicing-pipelines-nf/environment.yml
@@ -6,13 +6,13 @@ channels:
dependencies:
- fastqc=0.11.9
- trimmomatic=0.39
- - star=2.7.3
- - samtools=1.10
- - deeptools=3.4.0
- - multiqc=1.8
- - gffread=0.11.7
- - bioconductor-rtracklayer=1.46.0
+ - star=2.7.9a
+ - samtools=1.13
+ - deeptools=3.5.1
+ - multiqc=1.11
+ - bioconductor-rtracklayer=1.52.0
# Now installed via executable in `Dockerfile`
- # - stringtie=2.1.2
- # - gffcompare=0.11.2
\ No newline at end of file
+ # - stringtie=2.1.7
+ # - gffread=0.12.7 (version not available on conda)
+ # - gffcompare=0.12.6
\ No newline at end of file
diff --git a/nextflow.config b/nextflow.config
index f858a6e3..e23aec63 100755
--- a/nextflow.config
+++ b/nextflow.config
@@ -69,30 +69,30 @@ cleanup = params.cleanup
process {
errorStrategy = 'finish'
- container = 'gcr.io/nextflow-250616/splicing-pipelines-nf:gawk'
+ container = 'anczukowlab/splicing-pipelines-nf:3.0'
withName: 'get_accession' {
- container = 'lifebitai/download_reads:latest'
+ container = 'anczukowlab/download_reads:2.0'
}
withName: 'gen3_drs_fasp' {
- container = 'quay.io/lifebitai/lifebit-ai-fasp:latest'
+ container = 'anczukowlab/lifebit-ai-fasp:v1.1'
}
withName: 'get_tcga_bams' {
- container = 'lifebitai/download_reads:latest'
+ container = 'anczukowlab/download_reads:2.0'
}
withName: 'bamtofastq' {
- container = 'lifebitai/download_reads:latest'
+ container = 'anczukowlab/download_reads:2.0'
}
withName: 'rmats' {
- container = 'lifebitai/splicing-rmats:4.1.1'
+ container = 'anczukowlab/splicing-rmats:4.1.1'
}
withName: 'paired_rmats' {
- container = 'lifebitai/splicing-rmats:4.1.1'
+ container = 'anczukowlab/splicing-rmats:4.1.1'
}
withName: 'collect_tool_versions_env1' {
- container = 'gcr.io/nextflow-250616/splicing-pipelines-nf:gawk'
+ container = 'anczukowlab/splicing-pipelines-nf:3.0'
}
withName: 'collect_tool_versions_env2' {
- container = 'lifebitai/splicing-rmats:4.1.1'
+ container = 'anczukowlab/splicing-rmats:4.1.1'
}
}
From 3141ca177022b0399cf93f74f4e1bc3db9613d76 Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Fri, 13 Aug 2021 15:00:09 -0400
Subject: [PATCH 04/42] Update usage.md
Best solution to #246 is to update documentation.
---
docs/usage.md | 1 +
1 file changed, 1 insertion(+)
diff --git a/docs/usage.md b/docs/usage.md
index 862643c4..f330cd51 100755
--- a/docs/usage.md
+++ b/docs/usage.md
@@ -178,6 +178,7 @@ Other:
--skipMultiQC Skip MultiQC (bool)
(default: false)
--outdir The output directory where the results will be saved (string)
+ On Sumner, this must be set in the main.pbs. NF_splicing_pipeline.config will not overwrite main.pbs.
(default: directory where you submit the job)
--mega_time Sets time limit for processes withLabel 'mega_memory' in the main.nf using the base.config (time unit)
Make sure '#SBATCH -t' in 'main.pbs' is appropriately set if you are changing this parameter.
From 33e5d8aa1693be661a0074a101232510aeeca422 Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Fri, 13 Aug 2021 15:12:15 -0400
Subject: [PATCH 05/42] Update usage.md
---
docs/usage.md | 5 ++---
1 file changed, 2 insertions(+), 3 deletions(-)
diff --git a/docs/usage.md b/docs/usage.md
index f330cd51..fefdd545 100755
--- a/docs/usage.md
+++ b/docs/usage.md
@@ -117,6 +117,7 @@ Main arguments:
(default: false)
--stranded Specifies that the input is stranded ('first-strand', 'second-strand', false (aka unstranded))
(default: 'first-strand')
+ 'first-strand' refers to RF/fr-firststrand in this pipeline.
--readlength Read length - Note that all reads will be cropped to this length(int)
(default: no read length specified)
-profile Configuration profile to use. Can use multiple (comma separated, string)
@@ -239,7 +240,5 @@ Here `query_list.csv` should look something like -
```csv
file_name,sequencing_assay,data_format,file_name,sample_id,participant_id,tissue,age,gender
-GTEX-11EM3-1326-SM-5N9C6,RNA-Seq,bam,GTEX-11EM3-1326-SM-5N9C6.Aligned.sortedByCoord.out.patched.md.bam,GTEX-11EM3-1326-SM-5N9C6,GTEX-11EM3,Breast,21,Female
-GTEX-RU1J-0626-SM-4WAWY,RNA-Seq,bam,GTEX-RU1J-0626-SM-4WAWY.Aligned.sortedByCoord.out.patched.md.bam,GTEX-RU1J-0626-SM-4WAWY,GTEX-RU1J,Breast,21,Female
-GTEX-ZTPG-2826-SM-57WGA,RNA-Seq,bam,GTEX-ZTPG-2826-SM-57WGA.Aligned.sortedByCoord.out.patched.md.bam,GTEX-ZTPG-2826-SM-57WGA,GTEX-ZTPG,Breast,21,Female
+GTEX-PPPP-XXX-XX-XXXXX,RNA-Seq,bam,GTEX-PPPP-XXX-XX-XXXXX.Aligned.sortedByCoord.out.patched.md.bam,GTEX-PPPP-XXX-XX-XXXXX,GTEX-PPPP,Breast,21,Female
```
From 483e1f0d0e1810fefb2500837b9f26bc53d700d9 Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Fri, 13 Aug 2021 15:27:21 -0400
Subject: [PATCH 06/42] Update run_on_sumner.md
Adding some clarification to address #247
---
docs/run_on_sumner.md | 9 ++++++---
1 file changed, 6 insertions(+), 3 deletions(-)
diff --git a/docs/run_on_sumner.md b/docs/run_on_sumner.md
index a3c206a1..9c359211 100755
--- a/docs/run_on_sumner.md
+++ b/docs/run_on_sumner.md
@@ -19,6 +19,7 @@ This pipeline can be run on Sumner in three ways:
2. Input a 'reads' file to input fastq files and run pipeline until STAR mapping step with 'test' parameter.
3. Input a 'bams' file to input bams and run steps of pipeline following STAR mapping (Stringtie and rMATS).
+The `cacheDir` stores singularity images. This is set in splicing-pipelines-nf/conf/executors/sumner.config. For non-Anczukow users, this should be changed to a home directory.
## Running full pipeline with FASTQ input
### 1. Create a new run directory
@@ -81,14 +82,16 @@ If you already created a `NF_splicing_pipeline.config` during the trim test, you
- Each time you run the pipeline, go through all possible parameters to ensure you are creating a config ideal for your data. If you do not specify a value for a parameter, the default will be used. All parameters used can be found in the `log` file. WHEN IN DOUBT, SPECIFY ALL PARAMETERS
-- You must name your config file `NF_splicing_pipeline.config`
+- You must name your config file `NF_splicing_pipeline.config` (as specified in main.pbs)
- Your `NF_splicing_pipeline.config` must be in the directory that you are running your analysis.
-- The `readlength` here should be the length of the reads - if read leangth is not a multiple of 5 (ex- 76 or 151), set 'readlength' to nearest multiple of 5 (ex- 75 or 150). This extra base is an artifact of Illumina sequencing
+- The `readlength` here should be the length of the reads - if read length is not a multiple of 5 (ex- 76 or 151), set 'readlength' to nearest multiple of 5 (ex- 75 or 150). This extra base is an artifact of Illumina sequencing
- To run full pipeline, you **must** specify the following: `reads.csv`, `rmats_pairs.txt`, `readlength`, `assembly_name`, `star_index`, and `reference gtf`. This string can be a relative path from the directory in which you run Nextflow in, an absolute path or a link.
+- The star indexes must be generated prior to executing the pipeline (this is a separate step).
+
- Currently, the two options for genomes are hg38 and mm10. If you wish to use a newer version of the genome, you will need to add this to the post-processing script.
### 5. Run the pipeline!
@@ -106,7 +109,7 @@ Whereas parameters are set on the command-line using double dash options eg `--r
You can see some of these options [here](https://www.nextflow.io/docs/latest/tracing.html) in the Nextflow documentation.
-Some useful ones include:
+Some useful ones include (specified in main.pbs):
- `-resume` which will [resume](https://www.nextflow.io/docs/latest/getstarted.html?highlight=resume#modify-and-resume) any cached processes that have not been changed
- `-with-trace` eg `-with-trace trace.txt` which gives a [trace report](https://www.nextflow.io/docs/latest/tracing.html?highlight=dag#trace-report) for resource consumption by the pipeline
- `-with-dag` eg `-with-dag flowchart.png` which produces the [DAG visualisation](https://www.nextflow.io/docs/latest/tracing.html?highlight=dag#dag-visualisation) graph showing each of the different processes and the connections between them (the channels)
From 9027151f1c1ffef03afd3cd0f447c83d5859ad62 Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Fri, 13 Aug 2021 15:58:44 -0400
Subject: [PATCH 07/42] Update run_on_sumner.md
Adding the description to run pipeline with bams.csv to documentation
---
docs/run_on_sumner.md | 84 +++++++++++++++++++++++++++++++++++++++----
1 file changed, 78 insertions(+), 6 deletions(-)
diff --git a/docs/run_on_sumner.md b/docs/run_on_sumner.md
index 9c359211..91d6a9f5 100755
--- a/docs/run_on_sumner.md
+++ b/docs/run_on_sumner.md
@@ -15,9 +15,9 @@ git pull
Note: if you have not successfully completed the pipeline test, see [here](../README.md##quick-start-on-sumner-jaxs-hpc)
This pipeline can be run on Sumner in three ways:
- 1. Input a 'reads' file to input fastq files and run the pipeline in its entirety.
- 2. Input a 'reads' file to input fastq files and run pipeline until STAR mapping step with 'test' parameter.
- 3. Input a 'bams' file to input bams and run steps of pipeline following STAR mapping (Stringtie and rMATS).
+ 1. Input a `reads.csv` file to input fastq files and run the pipeline in its entirety.
+ 2. Input a `reads.csv` file to input fastq files and run pipeline until STAR mapping step with `--test` parameter set to `true`.
+ 3. Input a `bams.csv` file to input bams and run steps of pipeline following STAR mapping (Stringtie and rMATS).
The `cacheDir` stores singularity images. This is set in splicing-pipelines-nf/conf/executors/sumner.config. For non-Anczukow users, this should be changed to a home directory.
@@ -65,7 +65,7 @@ Each rMATS comparison must be specified with a comparison name as well as the `s
comparison2_id[space]sample3replicate1,sample3replicate2,sample3replicate3[space]sample4replicate1,sample4replicate1,sample4replicate1
```
- #### B1 only, no rMATS comparison (if this is run, set '--Statoff' parameter to 'true'):
+ #### B1 only, no rMATS comparison (if this is run, set '--statoff' parameter to 'true'):
```
comparison_id[space]sample1,sample2,sample3
```
@@ -76,11 +76,11 @@ Each rMATS comparison must be specified with a comparison name as well as the `s
This config file will be specific to your user and analysis. **You do not need to edit the pipeline code to configure the pipeline**. Descriptions of all possible parameters and their default values can be found [here](usage.md#all-available-parameters).
-If you already created a `NF_splicing_pipeline.config` during the trim test, you can modify it. Otherwise, to create your own custom config (to specify your input parameters) you can copy and edit this [example config](../conf/examples/MYC_MCF10A_0h_vs_MYC_MCF10A_8h.config) file.
+To create your own custom config (to specify your input parameters) you can copy and edit this [example config](../conf/examples/MYC_MCF10A_0h_vs_MYC_MCF10A_8h.config) file.
**VERY IMPORTANT NOTES***
-- Each time you run the pipeline, go through all possible parameters to ensure you are creating a config ideal for your data. If you do not specify a value for a parameter, the default will be used. All parameters used can be found in the `log` file. WHEN IN DOUBT, SPECIFY ALL PARAMETERS
+- Each time you run the pipeline, go through all possible parameters to ensure you are creating a config ideal for your data. If you do not specify a value for a parameter, the default will be used. All parameters used can be found in the `log` file. WHEN IN DOUBT, SPECIFY ALL PARAMETERS!
- You must name your config file `NF_splicing_pipeline.config` (as specified in main.pbs)
@@ -103,6 +103,78 @@ Run the pipeline!
sbatch /projects/anczukow-lab/splicing_pipeline/splicing-pipelines-nf/main.pbs
```
+## Running Stringtie and rMATS with BAM input
+### 1. Create a new run directory
+
+Create a new run directory within the appropriate dataset directory with the following format: runNumber_initials_date `run1_LU_20200519` (Example RUN Directory - `/projects/anczukow-lab/NGS_analysis/Dataset_4_MYC_MCF10A/run1_LU_20200519`).
+
+### 2. Create/Locate `bams.csv` file for your dataset
+
+Input reads are specified by the `bams` input parameter, specifying a path to a CSV file.
+
+- (create example) must contain columns for `sample_id`, `bam`, and `bam.bai`
+
+The 'bams.csv' column names must match the above example. The `sample_id` can be anything, however each must be unique. The `bam` column should contain the path to BAM files. The `bam.bai` column should contain the path to BAM.BAI files. You can create this on your local computer in excel and use WinSCP to move it to Sumner, or use create it using `nano` on the cluster.
+
+Supplying the `bams.csv` will signal to the pipeline to skip the first steps of the pipeline and start with Stringtie. No other parameter is needed.
+
+### 3. Create `rmats_pairs.txt` input file
+
+Each rMATS comparison must be specified with a comparison name as well as the `sample_id` as specified in the [`bams.csv`](create example) file. See example [`rmats_pairs.txt`](../examples/human_test/rmats_pairs.txt). Each line in the file corresponds to an rMATS execution. The first column corresponds to a unique name/id for the rMATS comparison (this will be used for the output folder/file names).
+
+* Replicates should be comma separated and the samples for the `b1` / `b2` files i.e. case and control should be space separated
+
+ See examples
+
+ #### Single sample pair:
+ ```
+ comparison_id[space]sample1[space]sample2
+ ```
+
+ #### Multiple sample pairs, no replicates:
+ ```
+ comparison1_id[space]sample1[space]sample2
+ comparison2_id[space]sample3[space]sample4
+ ```
+
+ #### Multiple sample pairs, with multiple replicates:
+ ```
+ comparison1_id[space]sample1replicate1,sample1replicate2,sample1replicate3[space]sample2replicate1,sample2replicate2,sample2replicate3
+ comparison2_id[space]sample3replicate1,sample3replicate2,sample3replicate3[space]sample4replicate1,sample4replicate1,sample4replicate1
+ ```
+
+ #### B1 only, no rMATS comparison (if this is run, set '--Statoff' parameter to 'true'):
+ ```
+ comparison_id[space]sample1,sample2,sample3
+ ```
+
+
+
+### 4. Setup `NF_splicing_pipeline.config`
+
+This config file will be specific to your user and analysis. **You do not need to edit the pipeline code to configure the pipeline**. Descriptions of all possible parameters and their default values can be found [here](usage.md#all-available-parameters).
+
+**VERY IMPORTANT NOTES***
+
+- Each time you run the pipeline, go through all possible parameters to ensure you are creating a config ideal for your data. If you do not specify a value for a parameter, the default will be used. All parameters used can be found in the `log` file. WHEN IN DOUBT, SPECIFY ALL PARAMETERS!
+
+- You must name your config file `NF_splicing_pipeline.config` (as specified in main.pbs).
+
+- Your `NF_splicing_pipeline.config` must be in the directory that you are running your analysis.
+
+- The `readlength` here should be the length of the reads - if read length is not a multiple of 5 (ex- 76 or 151), set 'readlength' to nearest multiple of 5 (ex- 75 or 150). This extra base is an artifact of Illumina sequencing
+
+- Currently, the two options for genomes are hg38 and mm10. If you wish to use a newer version of the genome, you will need to add this to the post-processing script.
+
+### 5. Run the pipeline!
+
+Ensure you have `NF_splicing_pipeline.config` in this directory.
+
+Run the pipeline!
+```
+sbatch /projects/anczukow-lab/splicing_pipeline/splicing-pipelines-nf/main.pbs
+```
+
# Bonus: useful Nextflow options
Whereas parameters are set on the command-line using double dash options eg `--reads`, parameters passed to Nextflow itself can be provided with single-dash options eg `-profile`.
From 2adb3362b90f54426b71ffcb9f0848efb2ac1064 Mon Sep 17 00:00:00 2001
From: cgpu <38183826+cgpu@users.noreply.github.com>
Date: Tue, 31 Aug 2021 17:54:17 +0300
Subject: [PATCH 08/42] Implements .command.* save in results/ (#251)
MIME-Version: 1.0
Content-Type: text/plain; charset=UTF-8
Content-Transfer-Encoding: 8bit
* Implements .command.* save in results/
* Cherry-pick residue removal
* Updates ci strategy to fail-fast: false
* Fix for "Unknown method `optional` on FileInParam type"
The mistake was I pasted in the input instead of the output directive
The failed ci is here:
https://github.com/TheJacksonLaboratory/splicing-pipelines-nf/pull/251/checks?check_run_id=3419423845#step:5:51
* Adds ${task.process} in command-log results naming
* Adds publishDir pattern: negation for logs in all results
* Adds tree view step for verifying results
* Removes install tree [already available] [docker]
* Removes install tree [already available] [singularity]
* Improves folder structure per sample for logs
* Keeps only .command.log, .command.sh, .command.err
* Adds config param (CloudOS configs)
* Removes redundancy of .bw files in star_mapped folder
After feedback from @angarb who spotted this redundancy,
we are removing the .bw file from the ${SRR}/ folder
and keeping it only in ${SRR}//all_bigwig only
├── star_mapped
│ ├── SRR4238351
│ │ ├── SRR4238351.Aligned.sortedByCoord.out.bam
│ │ ├── SRR4238351.Aligned.sortedByCoord.out.bam.bai
│ │ ├── SRR4238351.Log.final.out
│ │ ├── SRR4238351.Log.out
│ │ ├── SRR4238351.Log.progress.out
│ │ ├── SRR4238351.ReadsPerGene.out.tab
│ │ ├── SRR4238351.SJ.out.tab
│ │ ├── SRR4238351.Unmapped.out.mate1
│ │ └── SRR4238351.bw
│ └── all_bigwig
│ ├── SRR4238351.bw
* Sets fail-fast strategy to false in singularity CI
Complementary commit of https://github.com/TheJacksonLaboratory/splicing-pipelines-nf/pull/251/commits/09b8787b02da9622492d00b703e4b27e68ee685c
* Fix for indentation
---
.github/workflows/ci.yml | 4 ++
main.nf | 111 ++++++++++++++++++++++++++++++++++-----
nextflow.config | 8 +--
3 files changed, 106 insertions(+), 17 deletions(-)
diff --git a/.github/workflows/ci.yml b/.github/workflows/ci.yml
index 4f670fae..e2bb28ed 100755
--- a/.github/workflows/ci.yml
+++ b/.github/workflows/ci.yml
@@ -6,6 +6,7 @@ jobs:
docker:
runs-on: ubuntu-latest
strategy:
+ fail-fast: false
matrix:
nxf_ver: ['20.01.0', '']
steps:
@@ -18,9 +19,11 @@ jobs:
- name: Basic workflow tests
run: |
nextflow run ${GITHUB_WORKSPACE} -profile base,ultra_quick_test,docker
+ echo "Results tree view:" ; tree -a results
singularity:
runs-on: ubuntu-latest
strategy:
+ fail-fast: false
matrix:
singularity_version: ['3.6.4']
nxf_ver: ['20.01.0', '']
@@ -37,3 +40,4 @@ jobs:
- name: Basic workflow tests
run: |
nextflow run ${GITHUB_WORKSPACE} -profile base,ultra_quick_test,singularity
+ echo "Results tree view:" ; tree -a results
diff --git a/main.nf b/main.nf
index b2e32c7c..d11cf670 100755
--- a/main.nf
+++ b/main.nf
@@ -310,6 +310,8 @@ if ( download_from('gen3-drs')) {
if ( download_from('gtex') || download_from('sra') ) {
process get_accession {
+ publishDir "${params.outdir}/process-logs/${task.process}/${accession}/", pattern: "command-logs-*", mode: 'copy'
+
tag "${accession}"
label 'tiny_memory'
@@ -319,6 +321,7 @@ if ( download_from('gtex') || download_from('sra') ) {
output:
set val(accession), file(output_filename), val(params.singleEnd) into raw_reads_fastqc, raw_reads_trimmomatic
+ file("command-logs-*") optional true
script:
def ngc_cmd_with_key_file = key_file.name != 'no_key_file.txt' ? "--ngc ${key_file}" : ''
@@ -327,6 +330,9 @@ if ( download_from('gtex') || download_from('sra') ) {
prefetch $ngc_cmd_with_key_file $accession --progress -o $accession
fasterq-dump $ngc_cmd_with_key_file $accession --threads ${task.cpus} --split-3
pigz *.fastq
+
+ # save .command.* logs
+ ${params.savescript}
"""
}
}
@@ -339,7 +345,8 @@ if ( download_from('gen3-drs')) {
process gen3_drs_fasp {
tag "${file_name}"
label 'low_memory'
-
+ publishDir "${params.outdir}/process-logs/${task.process}/${file_name.baseName}", pattern: "command-logs-*", mode: 'copy'
+
input:
set val(subj_id), val(file_name), val(md5sum), val(obj_id), val(file_size) from ch_gtex_gen3_ids
each file(key_file) from key_file
@@ -347,6 +354,7 @@ if ( download_from('gen3-drs')) {
output:
set env(sample_name), file("*.bam"), val(false) into bamtofastq
+ file("command-logs-*") optional true
script:
"""
@@ -367,6 +375,9 @@ if ( download_from('gen3-drs')) {
if [[ ! "\$file_md5sum" =~ ${md5sum} ]]; then exit 1; else echo "file is good"; fi
samtools view -b -T ${genome_fasta} -o \${sample_name}.bam \${sample_name}.cram
fi
+
+ # save .command.* logs
+ ${params.savescript}
"""
}
}
@@ -379,6 +390,7 @@ if (download_from('tcga')) {
process get_tcga_bams {
tag "${accession}"
label 'low_memory'
+ publishDir "${params.outdir}/process-logs/${task.process}/", pattern: "command-logs-*", mode: 'copy'
input:
val(accession) from accession_ids
@@ -387,6 +399,7 @@ if (download_from('tcga')) {
output:
set val(accession), file("*.bam"), env(singleEnd) into bamtofastq
file("${accession}_paired_info.csv") into paired_info
+ file("command-logs-*") optional true
script:
// TODO: improve download speed by using `-n N_CONNECTIONS`
@@ -407,6 +420,9 @@ if (download_from('tcga')) {
echo "sample_id,n_single_reads,n_paired_reads,single_end" > ${accession}_paired_info.csv
echo "$accession,\$n_single_reads,\$n_paired_reads,\$singleEnd" >> ${accession}_paired_info.csv
+
+ # save .command.* logs
+ ${params.savescript}
"""
}
@@ -422,12 +438,14 @@ if (download_from('tcga') || download_from('gen3-drs')) {
process bamtofastq {
tag "${name}"
label 'mid_memory'
-
+ publishDir "${params.outdir}/process-logs/${task.process}/${name}/", pattern: "command-logs-*", mode: 'copy'
+
input:
set val(name), file(bam), val(singleEnd) from bamtofastq
output:
set val(name), file("*.fastq.gz"), val(singleEnd) into raw_reads_fastqc, raw_reads_trimmomatic
+ file("command-logs-*") optional true
script:
// samtools takes memory per thread
@@ -440,6 +458,9 @@ if (download_from('tcga') || download_from('gen3-drs')) {
"""
bedtools bamtofastq -i $bam -fq ${name}.fastq
pigz *.fastq
+
+ # save .command.* logs
+ ${params.savescript}
"""
} else {
"""
@@ -449,6 +470,9 @@ if (download_from('tcga') || download_from('gen3-drs')) {
-fq ${name}_1.fastq \
-fq2 ${name}_2.fastq
pigz *.fastq
+
+ # save .command.* logs
+ ${params.savescript}
"""
}
}
@@ -463,17 +487,22 @@ if (!params.bams){
process fastqc {
tag "$name"
label 'low_memory'
- publishDir "${params.outdir}/QC/raw", mode: 'copy'
+ publishDir "${params.outdir}/QC/raw", pattern: "*_fastqc.{zip,html}", mode: 'copy'
+ publishDir "${params.outdir}/process-logs/${task.process}/${name}", pattern: "command-logs-*", mode: 'copy'
input:
set val(name), file(reads), val(singleEnd) from raw_reads_fastqc
output:
file "*_fastqc.{zip,html}" into fastqc_results_raw
+ file("command-logs-*") optional true
script:
"""
fastqc --casava --threads $task.cpus $reads
+
+ # save .command.* logs
+ ${params.savescript}
"""
}
@@ -491,6 +520,7 @@ if (!params.bams){
process trimmomatic {
tag "$name"
label 'low_memory'
+ publishDir "${params.outdir}/process-logs/${task.process}/${name}", pattern: "command-logs-*", mode: 'copy'
input:
set val(name), file(reads), val(singleEnd), file(adapter) from raw_reads_trimmomatic_adapter
@@ -498,6 +528,7 @@ if (!params.bams){
output:
set val(name), file(output_filename), val(singleEnd) into (trimmed_reads_fastqc, trimmed_reads_star)
file ("logs/${name}_trimmomatic.log") into trimmomatic_logs
+ file("command-logs-*") optional true
script:
mode = singleEnd ? 'SE' : 'PE'
@@ -521,6 +552,9 @@ if (!params.bams){
mkdir logs
cp .command.log logs/${name}_trimmomatic.log
+
+ # save .command.* logs
+ ${params.savescript}
"""
}
@@ -531,17 +565,22 @@ if (!params.bams){
process fastqc_trimmed {
tag "$name"
label 'low_memory'
- publishDir "${params.outdir}/QC/trimmed", mode: 'copy'
+ publishDir "${params.outdir}/QC/trimmed", pattern: "*_fastqc.{zip,html}", mode: 'copy'
+ publishDir "${params.outdir}/process-logs/${task.process}/${name}", pattern: "command-logs-*", mode: 'copy'
input:
set val(name), file(reads), val(singleEnd) from trimmed_reads_fastqc
output:
file "*_fastqc.{zip,html}" into fastqc_results_trimmed
+ file("command-logs-*") optional true
script:
"""
fastqc --casava --threads $task.cpus $reads
+
+ # save .command.* logs
+ ${params.savescript}
"""
}
@@ -578,7 +617,8 @@ if (!params.bams){
process star {
tag "$name"
label 'mega_memory'
- publishDir "${params.outdir}/star_mapped/${name}", mode: 'copy'
+ publishDir "${params.outdir}/process-logs/${task.process}/${name}", pattern: "command-logs-*", mode: 'copy'
+ publishDir "${params.outdir}/star_mapped/${name}", pattern: "*{out.bam,out.bam.bai,out,ReadsPerGene.out.tab,SJ.out.tab,Unmapped}*" , mode: 'copy'
publishDir "${params.outdir}/star_mapped/", mode: 'copy',
saveAs: {filename ->
if (filename.indexOf(".bw") > 0) "all_bigwig/${name}.bw"
@@ -597,6 +637,7 @@ if (!params.bams){
file "*Log.out" into star_log
file "*Unmapped*" optional true
file "${name}.bw"
+ file("command-logs-*") optional true
script:
// TODO: check when to use `--outWigType wiggle` - for paired-end stranded stranded only?
@@ -647,8 +688,11 @@ if (!params.bams){
$xs_tag_cmd
samtools index ${name}.Aligned.sortedByCoord.out.bam
bamCoverage -b ${name}.Aligned.sortedByCoord.out.bam -o ${name}.bw
-
+
${post_script_run_resource_status}
+
+ # save .command.* logs
+ ${params.savescript}
"""
}
}
@@ -662,7 +706,8 @@ if (!params.test) {
process stringtie {
tag "$name"
label 'mega_memory'
- publishDir "${params.outdir}/star_mapped/${name}", mode: 'copy'
+ publishDir "${params.outdir}/star_mapped/${name}", pattern: "[!command-logs-]*", mode: 'copy'
+ publishDir "${params.outdir}/process-logs/${task.process}/${name}", pattern: "command-logs-*", mode: 'copy'
input:
set val(name), file(bam), file(bam_index) from indexed_bam
@@ -671,12 +716,16 @@ if (!params.test) {
output:
file "${name}.gtf" into stringtie_gtf
file "${name}_for_DGE.gtf" into stringtie_dge_gtf
+ file("command-logs-*") optional true
script:
rf = params.stranded ? params.stranded == 'first-strand' ? '--rf' : '--fr' : ''
"""
stringtie $bam -G $gtf -o ${name}.gtf $rf -a 8 -p $task.cpus
stringtie $bam -G $gtf -o ${name}_for_DGE.gtf $rf -a 8 -e -p $task.cpus
+
+ # save .command.* logs
+ ${params.savescript}
"""
}
@@ -686,10 +735,12 @@ if (!params.test) {
process prep_de {
label 'mid_memory'
- publishDir "${params.outdir}/star_mapped/count_matrix", mode: 'copy'
+ publishDir "${params.outdir}/star_mapped/count_matrix", pattern: "[!command-logs-]*", mode: 'copy'
+ publishDir "${params.outdir}/process-logs/${task.process}/", pattern: "command-logs-*", mode: 'copy'
input:
file(gtf) from stringtie_dge_gtf.collect()
+ file("command-logs-*") optional true
output:
file "sample_lst.txt"
@@ -703,6 +754,9 @@ if (!params.test) {
paste -d ' ' samples.txt gtfs.txt > sample_lst.txt
prepDE.py -i sample_lst.txt -l $params.readlength \
-g ${run_prefix}_gene_count_matrix.csv -t ${run_prefix}_transcript_count_matrix.csv
+
+ # save .command.* logs
+ ${params.savescript}
"""
}
@@ -712,7 +766,8 @@ if (!params.test) {
process stringtie_merge {
label 'mid_memory'
- publishDir "${params.outdir}/star_mapped/stringtie_merge", mode: 'copy'
+ publishDir "${params.outdir}/star_mapped/stringtie_merge", pattern: "[!command-logs-]*", mode: 'copy'
+ publishDir "${params.outdir}/process-logs/${task.process}/", pattern: "command-logs-*", mode: 'copy'
input:
file('*.gtf') from stringtie_gtf.collect()
@@ -721,6 +776,7 @@ if (!params.test) {
output:
file "gffcmp.annotated.corrected.gtf" into merged_gtf
file "gffcmp.*" into gffcmp
+ file("command-logs-*") optional true
script:
"""
@@ -729,6 +785,9 @@ if (!params.test) {
gffcompare -R -V -r $gtf stringtie_merged.gtf
correct_gene_names.R
gffread -E gffcmp.annotated.corrected.gff -T -o gffcmp.annotated.corrected.gtf
+
+ # save .command.* logs
+ ${params.savescript}
"""
}
@@ -777,7 +836,8 @@ if (!params.test) {
process rmats {
tag "$rmats_id ${gtf.simpleName}"
label 'high_memory'
- publishDir "${params.outdir}/rMATS_out/${rmats_id}_${gtf.simpleName}", mode: 'copy'
+ publishDir "${params.outdir}/rMATS_out/${rmats_id}_${gtf.simpleName}", pattern: "[!command-logs-]*", mode: 'copy'
+ publishDir "${params.outdir}/process-logs/${task.process}/${rmats_id}_${gtf.simpleName}", pattern: "command-logs-*", mode: 'copy'
when:
!params.skiprMATS
@@ -788,6 +848,7 @@ if (!params.test) {
output:
file "*.{txt,csv}" into rmats_out
+ file("command-logs-*") optional true
script:
libType = params.stranded ? params.stranded == 'first-strand' ? 'fr-firststrand' : 'fr-secondstrand' : 'fr-unstranded'
@@ -834,6 +895,9 @@ if (!params.test) {
echo rmats_id ${rmats_id} >> \$rmats_config
LU_postprocessing.R
+
+ # save .command.* logs
+ ${params.savescript}
"""
}
@@ -849,7 +913,8 @@ if (!params.test) {
process paired_rmats {
tag "$name1 $name2"
label 'high_memory'
- publishDir "${params.outdir}/rMATS_out/${name1}_vs_${name2}_${gtf.simpleName}", mode: 'copy'
+ publishDir "${params.outdir}/rMATS_out/${name1}_vs_${name2}_${gtf.simpleName}", pattern: "[!command-logs-]*", mode: 'copy'
+ publishDir "${params.outdir}/process-logs/${task.process}/${name1}_vs_${name2}_${gtf.simpleName}", pattern: "command-logs-*", mode: 'copy'
when:
!params.skiprMATS
@@ -860,6 +925,7 @@ if (!params.test) {
output:
file "*.{txt,csv}" into paired_rmats_out
+ file("command-logs-*") optional true
script:
libType = params.stranded ? params.stranded == 'first-strand' ? 'fr-firststrand' : 'fr-secondstrand' : 'fr-unstranded'
@@ -895,6 +961,9 @@ if (!params.test) {
echo rmats_id ${name1}_vs_${name2} >> \$rmats_config
LU_postprocessing.R
+
+ # save .command.* logs
+ ${params.savescript}
"""
}
}
@@ -907,7 +976,8 @@ if (!params.test) {
if (!params.bams) {
process multiqc {
label 'mega_memory'
- publishDir "${params.outdir}/MultiQC", mode: 'copy'
+ publishDir "${params.outdir}/MultiQC", pattern: "[!command-logs-]*", mode: 'copy'
+ publishDir "${params.outdir}/process-logs/${task.process}/", pattern: "command-logs-*", mode: 'copy'
when:
!params.skipMultiQC
@@ -923,11 +993,15 @@ if (!params.bams) {
file "*multiqc_report.html" into multiqc_report
file "*_data/*"
file ('trimmomatic')
+ file("command-logs-*") optional true
script:
"""
multiqc . --config $multiqc_config -m fastqc -m star -m trimmomatic
cp multiqc_report.html ${run_prefix}_multiqc_report.html
+
+ # save .command.* logs
+ ${params.savescript}
"""
}
}
@@ -938,9 +1012,11 @@ if (!params.bams) {
process collect_tool_versions_env1 {
// TODO: This collects tool versions for only one base enviroment/container - 'gcr.io/nextflow-250616/splicing-pipelines-nf:gawk'
// need to get tool versions from other enviroment/container
+ publishDir "${params.outdir}/process-logs/${task.process}/", pattern: "command-logs-*", mode: 'copy'
output:
file("tool_versions.txt") into ch_tool_versions
+ file("command-logs-*") optional true
script:
"""
@@ -953,23 +1029,30 @@ process collect_tool_versions_env1 {
conda list -n splicing-pipelines-nf | grep multiqc | tail -n 1 >> tool_versions.txt
conda list -n splicing-pipelines-nf | grep gffread | tail -n 1 >> tool_versions.txt
echo -e "stringtie" ' \t\t\t\t ' \$(stringtie --version) >> tool_versions.txt
+
+ # save .command.* logs
+ ${params.savescript}
"""
}
process collect_tool_versions_env2 {
echo true
- publishDir "${params.outdir}", mode: 'copy'
-
+ publishDir "${params.outdir}/tool-versions/env2/", pattern: "[!command-logs-]*", mode: 'copy'
+ publishDir "${params.outdir}/process-logs/${task.process}/", pattern: "command-logs-*", mode: 'copy'
input:
file(tool_versions) from ch_tool_versions
output:
file("tool_versions.txt") into ch_all_tool_versions
+ file("command-logs-*") optional true
script:
"""
conda list -n rmats4 | grep rmats | tail -n 1 >> tool_versions.txt
+
+ # save .command.* logs
+ ${params.savescript}
"""
}
diff --git a/nextflow.config b/nextflow.config
index e23aec63..c0eba41b 100755
--- a/nextflow.config
+++ b/nextflow.config
@@ -53,6 +53,9 @@ params {
tracedir = "${params.outdir}/pipeline_info"
cleanup = false // if true will delete all intermediate files in work folder on workflow completion (not including staged files)
+ // Save of .command.* logs
+ savescript = 'task_hash=`basename \${PWD} | cut -c1-6`; mkdir command-logs-\$task_hash ; cp .command.*{err,log,sh} command-logs-\$task_hash'
+
// Max resources
max_memory = 760.GB
max_cpus = 72
@@ -63,6 +66,7 @@ params {
// google life science specific
debug = false
gls_boot_disk_size = 50.GB
+ config = 'conf/executors/google.config'
}
cleanup = params.cleanup
@@ -97,9 +101,7 @@ process {
}
profiles {
- standard {
- includeConfig 'conf/executors/google.config'
- }
+ standard { includeConfig params.config }
docker {
docker.enabled = true
docker.runOptions = '-u $(id -u):$(id -g)' // to prevent files in workdir owned by root user
From 9464dbcf45742fa43c26682f1d3c4eaefaf1cab1 Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Wed, 15 Sep 2021 09:46:46 +0300
Subject: [PATCH 09/42] Fix saving files (#263)
* Fixes *_data/* files not being saved for multiqcs step
* Fixes sample_lst.txt not being saved for prep_de step
* Fixes no files being saved for stringtie_merge step
* Fix prep_de step input
* Saves tmp/*_read_outcomes_by_bam.txt in both rmats proc
---
main.nf | 14 ++++++++------
1 file changed, 8 insertions(+), 6 deletions(-)
diff --git a/main.nf b/main.nf
index d11cf670..9fb90136 100755
--- a/main.nf
+++ b/main.nf
@@ -735,17 +735,17 @@ if (!params.test) {
process prep_de {
label 'mid_memory'
- publishDir "${params.outdir}/star_mapped/count_matrix", pattern: "[!command-logs-]*", mode: 'copy'
+ publishDir "${params.outdir}/star_mapped/count_matrix", pattern: "{sample_lst.txt,*gene_count_matrix.csv,*transcript_count_matrix.csv}", mode: 'copy'
publishDir "${params.outdir}/process-logs/${task.process}/", pattern: "command-logs-*", mode: 'copy'
input:
file(gtf) from stringtie_dge_gtf.collect()
- file("command-logs-*") optional true
output:
file "sample_lst.txt"
file "*gene_count_matrix.csv"
file "*transcript_count_matrix.csv"
+ file("command-logs-*") optional true
script:
"""
@@ -766,7 +766,7 @@ if (!params.test) {
process stringtie_merge {
label 'mid_memory'
- publishDir "${params.outdir}/star_mapped/stringtie_merge", pattern: "[!command-logs-]*", mode: 'copy'
+ publishDir "${params.outdir}/star_mapped/stringtie_merge", pattern: "{gffcmp.annotated.corrected.gtf,gffcmp.*}", mode: 'copy'
publishDir "${params.outdir}/process-logs/${task.process}/", pattern: "command-logs-*", mode: 'copy'
input:
@@ -836,7 +836,7 @@ if (!params.test) {
process rmats {
tag "$rmats_id ${gtf.simpleName}"
label 'high_memory'
- publishDir "${params.outdir}/rMATS_out/${rmats_id}_${gtf.simpleName}", pattern: "[!command-logs-]*", mode: 'copy'
+ publishDir "${params.outdir}/rMATS_out/${rmats_id}_${gtf.simpleName}", pattern: "{*.txt,*.csv,tmp/*_read_outcomes_by_bam.txt}", mode: 'copy'
publishDir "${params.outdir}/process-logs/${task.process}/${rmats_id}_${gtf.simpleName}", pattern: "command-logs-*", mode: 'copy'
when:
@@ -848,6 +848,7 @@ if (!params.test) {
output:
file "*.{txt,csv}" into rmats_out
+ file "tmp/*_read_outcomes_by_bam.txt"
file("command-logs-*") optional true
script:
@@ -913,7 +914,7 @@ if (!params.test) {
process paired_rmats {
tag "$name1 $name2"
label 'high_memory'
- publishDir "${params.outdir}/rMATS_out/${name1}_vs_${name2}_${gtf.simpleName}", pattern: "[!command-logs-]*", mode: 'copy'
+ publishDir "${params.outdir}/rMATS_out/${name1}_vs_${name2}_${gtf.simpleName}", pattern: "{*.txt,*.csv,tmp/*_read_outcomes_by_bam.txt}", mode: 'copy'
publishDir "${params.outdir}/process-logs/${task.process}/${name1}_vs_${name2}_${gtf.simpleName}", pattern: "command-logs-*", mode: 'copy'
when:
@@ -925,6 +926,7 @@ if (!params.test) {
output:
file "*.{txt,csv}" into paired_rmats_out
+ file "tmp/*_read_outcomes_by_bam.txt"
file("command-logs-*") optional true
script:
@@ -976,7 +978,7 @@ if (!params.test) {
if (!params.bams) {
process multiqc {
label 'mega_memory'
- publishDir "${params.outdir}/MultiQC", pattern: "[!command-logs-]*", mode: 'copy'
+ publishDir "${params.outdir}/MultiQC", pattern: "{*multiqc_report.html,*_data/*,trimmomatic}", mode: 'copy'
publishDir "${params.outdir}/process-logs/${task.process}/", pattern: "command-logs-*", mode: 'copy'
when:
From ed4c6ac1b6e744f9e0f5569646958ed8f69d5578 Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Wed, 15 Sep 2021 10:34:48 +0300
Subject: [PATCH 10/42] Adds xstag strType parameter (#264)
---
main.nf | 3 ++-
nextflow.config | 8 ++++++++
2 files changed, 10 insertions(+), 1 deletion(-)
diff --git a/main.nf b/main.nf
index 9fb90136..216847ec 100755
--- a/main.nf
+++ b/main.nf
@@ -179,6 +179,7 @@ log.info "Single-end : ${download_from('tcga') ? 'Will be check
log.info "GTF : ${params.gtf}"
log.info "STAR index : ${star_index}"
log.info "Stranded : ${params.stranded}"
+log.info "strType : ${params.strType[params.stranded].strType}"
log.info "Soft_clipping : ${params.soft_clipping}"
log.info "rMATS pairs file : ${params.rmats_pairs ? params.rmats_pairs : 'Not provided'}"
log.info "Adapter : ${download_from('tcga') ? 'Will be set for each sample based based on whether the sample is paired or single-end' : adapter_file}"
@@ -644,7 +645,7 @@ if (!params.bams){
// TODO: find a better solution to needing to use `chmod`
out_filter_intron_motifs = params.stranded ? '' : '--outFilterIntronMotifs RemoveNoncanonicalUnannotated'
out_sam_strand_field = params.stranded ? '' : '--outSAMstrandField intronMotif'
- xs_tag_cmd = params.stranded ? "samtools view -h ${name}.Aligned.sortedByCoord.out.bam | gawk -v strType=2 -f /usr/local/bin/tagXSstrandedData.awk | samtools view -bS - > Aligned.XS.bam && mv Aligned.XS.bam ${name}.Aligned.sortedByCoord.out.bam" : ''
+ xs_tag_cmd = params.stranded ? "samtools view -h ${name}.Aligned.sortedByCoord.out.bam | gawk -v q=${params.strType[params.stranded].strType} -f /usr/local/bin/tagXSstrandedData.awk | samtools view -bS - > Aligned.XS.bam && mv Aligned.XS.bam ${name}.Aligned.sortedByCoord.out.bam" : ''
endsType = params.soft_clipping ? 'Local' : 'EndToEnd'
// Set maximum available memory to be used by STAR to sort BAM files
star_mem = params.star_memory ? params.star_memory : task.memory
diff --git a/nextflow.config b/nextflow.config
index c0eba41b..2d24389e 100755
--- a/nextflow.config
+++ b/nextflow.config
@@ -20,6 +20,14 @@ params {
star_index = false
singleEnd = false
stranded = 'first-strand'
+ strType {
+ 'first-strand' {
+ strType = 2
+ }
+ 'second-strand' {
+ strType = 1
+ }
+ }
readlength = false
// Trimmomatic:
From 265a24330427d4661ac8f2fdbfb1ba9e1257661f Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Wed, 15 Sep 2021 10:57:29 +0300
Subject: [PATCH 11/42] Adds sra test (#253)
* Adds SRA test profile
* Adds sra_test to ci tests
* Changes CI strategy to fail-fast:false
* CI syntax fix [previous commit]
* Parameterises echo in process scope
* Adds echo true for ci debugging
* Fixes sra-toolkit run with singularity
* Change sra example file to a really small one
* Revert the main container version to the newer one
* Removes commented unnecessary docker.runOptions line
* Removes failing sra_test ci test for singularity
* Fix star step in ci test
* Makes more robust star issue solution
* Returns errorStrategy = 'finish'
Co-authored-by: cgpu <38183826+cgpu@users.noreply.github.com>
---
.github/workflows/ci.yml | 8 +++++---
assets/sra-user-settings.mkfg | 2 ++
conf/examples/sra_test.config | 31 +++++++++++++++++++++++++++++++
examples/testdata/sra/sra.csv | 2 ++
main.nf | 11 +++++++++++
nextflow.config | 8 +++++++-
6 files changed, 58 insertions(+), 4 deletions(-)
create mode 100644 assets/sra-user-settings.mkfg
create mode 100755 conf/examples/sra_test.config
create mode 100644 examples/testdata/sra/sra.csv
diff --git a/.github/workflows/ci.yml b/.github/workflows/ci.yml
index e2bb28ed..ff33a593 100755
--- a/.github/workflows/ci.yml
+++ b/.github/workflows/ci.yml
@@ -9,6 +9,7 @@ jobs:
fail-fast: false
matrix:
nxf_ver: ['20.01.0', '']
+ test_type: ['ultra_quick_test', 'sra_test']
steps:
- uses: actions/checkout@v1
- name: Install Nextflow
@@ -18,7 +19,7 @@ jobs:
sudo mv nextflow /usr/local/bin/
- name: Basic workflow tests
run: |
- nextflow run ${GITHUB_WORKSPACE} -profile base,ultra_quick_test,docker
+ nextflow run ${GITHUB_WORKSPACE} -profile base,${{ matrix.test_type }},docker
echo "Results tree view:" ; tree -a results
singularity:
runs-on: ubuntu-latest
@@ -27,6 +28,7 @@ jobs:
matrix:
singularity_version: ['3.6.4']
nxf_ver: ['20.01.0', '']
+ test_type: ['ultra_quick_test']
steps:
- uses: actions/checkout@v1
- uses: eWaterCycle/setup-singularity@v6
@@ -39,5 +41,5 @@ jobs:
sudo mv nextflow /usr/local/bin/
- name: Basic workflow tests
run: |
- nextflow run ${GITHUB_WORKSPACE} -profile base,ultra_quick_test,singularity
- echo "Results tree view:" ; tree -a results
+ nextflow run ${GITHUB_WORKSPACE} -profile base,${{ matrix.test_type }},singularity --echo true
+ echo "Results tree view:" ; tree -a results
\ No newline at end of file
diff --git a/assets/sra-user-settings.mkfg b/assets/sra-user-settings.mkfg
new file mode 100644
index 00000000..1cef9b5c
--- /dev/null
+++ b/assets/sra-user-settings.mkfg
@@ -0,0 +1,2 @@
+/LIBS/IMAGE_GUID = "aee5f45c-f469-45f1-95f2-b2d2b1c59163"
+/libs/cloud/report_instance_identity = "true"
diff --git a/conf/examples/sra_test.config b/conf/examples/sra_test.config
new file mode 100755
index 00000000..b4cb9e64
--- /dev/null
+++ b/conf/examples/sra_test.config
@@ -0,0 +1,31 @@
+/*
+ * -------------------------------------------------
+ * Nextflow config file for running ultra quick tests
+ * -------------------------------------------------
+ * Defines bundled input files and everything required
+ * to run a fast and simple test. Use as follows:
+ * nextflow run jacksonlabs/splicing-pipelines-nf -profile ultra_quick_test
+ *
+ */
+
+params {
+ // Input data
+ singleEnd = true
+ reads = "$baseDir/examples/testdata/sra/sra.csv"
+ download_from = 'SRA'
+
+ // Genome references
+ gtf = 'https://lifebit-featured-datasets.s3-eu-west-1.amazonaws.com/projects/jax/splicing-pipelines-nf/genes.gtf'
+ star_index = 'https://lifebit-featured-datasets.s3-eu-west-1.amazonaws.com/projects/jax/splicing-pipelines-nf/star_2.7.9a_yeast_chr_I.tar.gz'
+
+ // Other
+ test = true
+ readlength = 500
+ // This doesn't make biological sense but prevents all reads being removed during trimming
+ overhang = 100
+
+ // Limit resources
+ max_cpus = 2
+ max_memory = 6.GB
+ max_time = 48.h
+}
diff --git a/examples/testdata/sra/sra.csv b/examples/testdata/sra/sra.csv
new file mode 100644
index 00000000..28bf315b
--- /dev/null
+++ b/examples/testdata/sra/sra.csv
@@ -0,0 +1,2 @@
+sample_id
+ERR4667735
diff --git a/main.nf b/main.nf
index 216847ec..46fa4f2e 100755
--- a/main.nf
+++ b/main.nf
@@ -305,6 +305,13 @@ if ( download_from('gen3-drs')) {
.set {ch_genome_fasta}
}
+if ( download_from('sra')) {
+ Channel
+ .value(file(params.sra_config_file))
+ .set {ch_sra_config_file}
+}
+
+
/*--------------------------------------------------
Download FASTQs from GTEx or SRA
---------------------------------------------------*/
@@ -319,6 +326,7 @@ if ( download_from('gtex') || download_from('sra') ) {
input:
val(accession) from accession_ids
each file(key_file) from key_file
+ file(sra_config) from ch_sra_config_file
output:
set val(accession), file(output_filename), val(params.singleEnd) into raw_reads_fastqc, raw_reads_trimmomatic
@@ -328,6 +336,8 @@ if ( download_from('gtex') || download_from('sra') ) {
def ngc_cmd_with_key_file = key_file.name != 'no_key_file.txt' ? "--ngc ${key_file}" : ''
output_filename = params.singleEnd ? "${accession}.fastq.gz" : "${accession}_{1,2}.fastq.gz"
"""
+ mkdir .ncbi
+ mv ${sra_config} .ncbi/
prefetch $ngc_cmd_with_key_file $accession --progress -o $accession
fasterq-dump $ngc_cmd_with_key_file $accession --threads ${task.cpus} --split-3
pigz *.fastq
@@ -691,6 +701,7 @@ if (!params.bams){
bamCoverage -b ${name}.Aligned.sortedByCoord.out.bam -o ${name}.bw
${post_script_run_resource_status}
+ rm -r ${file(index).name.minus('.gz').minus('.tar')} # not simpleName or twice baseName because index has dot's in name: star_2.7.9a_yeast_chr_I.tar.gz
# save .command.* logs
${params.savescript}
diff --git a/nextflow.config b/nextflow.config
index 2d24389e..6c2b8058 100755
--- a/nextflow.config
+++ b/nextflow.config
@@ -11,6 +11,7 @@ params {
rmats_pairs = false
run_name = false
download_from = false
+ sra_config_file= "${baseDir}/assets/sra-user-settings.mkfg"
key_file = false
genome_fasta = false
@@ -74,6 +75,10 @@ params {
// google life science specific
debug = false
gls_boot_disk_size = 50.GB
+
+ // process scope options
+ echo = false
+
config = 'conf/executors/google.config'
}
@@ -81,6 +86,7 @@ cleanup = params.cleanup
process {
errorStrategy = 'finish'
+ echo = params.echo
container = 'anczukowlab/splicing-pipelines-nf:3.0'
withName: 'get_accession' {
container = 'anczukowlab/download_reads:2.0'
@@ -112,7 +118,6 @@ profiles {
standard { includeConfig params.config }
docker {
docker.enabled = true
- docker.runOptions = '-u $(id -u):$(id -g)' // to prevent files in workdir owned by root user
}
base { includeConfig 'conf/executors/base.config' }
sumner {
@@ -124,6 +129,7 @@ profiles {
}
MYC_MCF10A_0h_vs_MYC_MCF10A_8h { includeConfig 'conf/examples/MYC_MCF10A_0h_vs_MYC_MCF10A_8h.config' }
ultra_quick_test { includeConfig 'conf/examples/ultra_quick_test.config' }
+ sra_test { includeConfig 'conf/examples/sra_test.config' }
}
dag {
From 7243f3740d34296dc91e3b8d6c79090cd151c781 Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Thu, 23 Sep 2021 19:19:09 +0300
Subject: [PATCH 12/42] Add b1 - control and b2- case to docs
---
docs/run_on_sumner.md | 2 +-
1 file changed, 1 insertion(+), 1 deletion(-)
diff --git a/docs/run_on_sumner.md b/docs/run_on_sumner.md
index 91d6a9f5..22716abd 100755
--- a/docs/run_on_sumner.md
+++ b/docs/run_on_sumner.md
@@ -44,7 +44,7 @@ There should be one `reads.csv` file per dataset. If your dataset already has a
Each rMATS comparison must be specified with a comparison name as well as the `sample_id` as specified in the [`reads`](../examples/testdata/human_test/human_test_reps.csv) file. See example [`rmats_pairs.txt`](../examples/human_test/rmats_pairs.txt). Each line in the file corresponds to an rMATS execution. The first column corresponds to a unique name/id for the rMATS comparison (this will be used for the output folder/file names)
-* Replicates should be comma separated and the samples for the `b1` / `b2` files i.e. case and control should be space separated
+* Replicates should be comma separated and the samples for the `b1` / `b2` files i.e. case and control should be space separated. b1 - control and b2 - case.
See examples
From 029d14b9f5ee41bd0532bffb15000ab14ebe2949 Mon Sep 17 00:00:00 2001
From: Brittany Angarola
Date: Mon, 27 Sep 2021 11:51:22 -0400
Subject: [PATCH 13/42] permissions changes
---
DAG.png | Bin
assets/sra-user-settings.mkfg | 0
conf/executors/singularity.config | 0
containers/fasp/Dockerfile | 2 +-
docs/Running_TCGA.md | 0
docs/run_locally.md | 0
examples/testdata/sra/sra.csv | 0
7 files changed, 1 insertion(+), 1 deletion(-)
mode change 100644 => 100755 DAG.png
mode change 100644 => 100755 assets/sra-user-settings.mkfg
mode change 100644 => 100755 conf/executors/singularity.config
mode change 100644 => 100755 docs/Running_TCGA.md
mode change 100644 => 100755 docs/run_locally.md
mode change 100644 => 100755 examples/testdata/sra/sra.csv
diff --git a/DAG.png b/DAG.png
old mode 100644
new mode 100755
diff --git a/assets/sra-user-settings.mkfg b/assets/sra-user-settings.mkfg
old mode 100644
new mode 100755
diff --git a/conf/executors/singularity.config b/conf/executors/singularity.config
old mode 100644
new mode 100755
diff --git a/containers/fasp/Dockerfile b/containers/fasp/Dockerfile
index 81f2d27f..55694785 100644
--- a/containers/fasp/Dockerfile
+++ b/containers/fasp/Dockerfile
@@ -17,7 +17,7 @@ RUN apt-get update \
&& cd fasp-scripts \
&& python setup.py install \
&& chmod +x fasp/scripts/* \
- && conda install samtools=1.13 -c bioconda -c conda-forge \
+ && conda install samtools=1.11 -c bioconda -c conda-forge \
&& conda clean -a
ENV PATH /fasp-scripts/fasp/scripts:$PATH
diff --git a/docs/Running_TCGA.md b/docs/Running_TCGA.md
old mode 100644
new mode 100755
diff --git a/docs/run_locally.md b/docs/run_locally.md
old mode 100644
new mode 100755
diff --git a/examples/testdata/sra/sra.csv b/examples/testdata/sra/sra.csv
old mode 100644
new mode 100755
From 76d541e46b47e38b5f942d687822fd0439309928 Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Mon, 27 Sep 2021 20:35:42 +0300
Subject: [PATCH 14/42] Fix strType issue when stranded=false (#276)
---
main.nf | 2 +-
1 file changed, 1 insertion(+), 1 deletion(-)
diff --git a/main.nf b/main.nf
index 46fa4f2e..7115b084 100755
--- a/main.nf
+++ b/main.nf
@@ -179,7 +179,7 @@ log.info "Single-end : ${download_from('tcga') ? 'Will be check
log.info "GTF : ${params.gtf}"
log.info "STAR index : ${star_index}"
log.info "Stranded : ${params.stranded}"
-log.info "strType : ${params.strType[params.stranded].strType}"
+if (params.stranded) {log.info "strType : ${params.strType[params.stranded].strType}"}
log.info "Soft_clipping : ${params.soft_clipping}"
log.info "rMATS pairs file : ${params.rmats_pairs ? params.rmats_pairs : 'Not provided'}"
log.info "Adapter : ${download_from('tcga') ? 'Will be set for each sample based based on whether the sample is paired or single-end' : adapter_file}"
From 10a2118c9195945a4dfa41927cb7d544e56905b9 Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Mon, 27 Sep 2021 13:45:12 -0400
Subject: [PATCH 15/42] Update Dockerfile
Accidentally edited this file.
---
containers/fasp/Dockerfile | 2 +-
1 file changed, 1 insertion(+), 1 deletion(-)
diff --git a/containers/fasp/Dockerfile b/containers/fasp/Dockerfile
index 55694785..81f2d27f 100644
--- a/containers/fasp/Dockerfile
+++ b/containers/fasp/Dockerfile
@@ -17,7 +17,7 @@ RUN apt-get update \
&& cd fasp-scripts \
&& python setup.py install \
&& chmod +x fasp/scripts/* \
- && conda install samtools=1.11 -c bioconda -c conda-forge \
+ && conda install samtools=1.13 -c bioconda -c conda-forge \
&& conda clean -a
ENV PATH /fasp-scripts/fasp/scripts:$PATH
From eedda346937871408939e366ba4360151bb9bec1 Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Mon, 27 Sep 2021 20:52:00 +0300
Subject: [PATCH 16/42] Parametrize error strategy (clean pr) (#267)
* Parametrizes error strategy
* Adds error_strategy parameter to usage.md docs
* Update log.info to show actual errorStrategy value
* Fix typo
---
docs/usage.md | 4 ++++
main.nf | 10 ++++++++++
nextflow.config | 6 +++++-
3 files changed, 19 insertions(+), 1 deletion(-)
diff --git a/docs/usage.md b/docs/usage.md
index fefdd545..dad6be73 100755
--- a/docs/usage.md
+++ b/docs/usage.md
@@ -189,6 +189,10 @@ Other:
--debug This option will enable echo of script execution into STDOUT with some additional
resource information (such as machine type, memory, cpu and disk space)
(default: false)
+ --error_strategy Mode of pipeline handling failed processes. Possible values: 'terminate', 'finish', 'ignore', 'retry'.
+ Check nextflow documnetation for detailed descriptions of each mode:
+ https://www.nextflow.io/docs/latest/process.html#process-page-error-strategy
+ (default: finish)
--cleanup This option will enable nextflow work folder cleanup upon pipeline successfull completion.
All intermediate files from nexftlow processes' workdirs will be cleared, staging folder with staged
files will not be cleared.
diff --git a/main.nf b/main.nf
index 7115b084..fff032c0 100755
--- a/main.nf
+++ b/main.nf
@@ -116,6 +116,10 @@ def helpMessage() {
--debug This option will enable echo of script execution into STDOUT with some additional
resource information (such as machine type, memory, cpu and disk space)
(default: false)
+ --error_strategy Mode of pipeline handling failed processes. Possible values: 'terminate', 'finish', 'ignore', 'retry'.
+ Check nextflow documnetation for detailed descriptions of each mode:
+ https://www.nextflow.io/docs/latest/process.html#process-page-error-strategy
+ (default: $params.error_strategy)
--cleanup This option will enable nextflow work folder cleanup upon pipeline successfull completion.
All intermediate files from nexftlow processes' workdirs will be cleared, staging folder with staged
files will not be cleared.
@@ -151,6 +155,11 @@ if (!params.bams) {
star_index = false
}
+// Check if error_strategy parameter has a correct value
+if (!params.allowed_error_strategies.contains(params.error_strategy)) {
+ exit 1, "Error strategy \"${params.error_strategy}\" is not correct. Please choose one of: ${params.allowed_error_strategies.join(", ")}."
+}
+
// Check if user has set adapter sequence. If not set is based on the value of the singleEnd parameter
adapter_file = params.adapter ? params.adapter : params.singleEnd ? "$baseDir/adapters/TruSeq3-SE.fa" : "$baseDir/adapters/TruSeq3-PE.fa"
// Set overhang to read length -1, unless user specified
@@ -208,6 +217,7 @@ log.info "Max time : ${params.max_time}"
log.info "Mega time : ${params.mega_time}"
log.info "Google Cloud disk-space : ${params.gc_disk_size}"
log.info "Debug : ${params.debug}"
+log.info "Error strategy : ${config.process.errorStrategy}"
log.info "Workdir cleanup : ${params.cleanup}"
log.info ""
log.info "\n"
diff --git a/nextflow.config b/nextflow.config
index 6c2b8058..d2e4c2ae 100755
--- a/nextflow.config
+++ b/nextflow.config
@@ -60,6 +60,8 @@ params {
help = false
mega_time = 20.h
tracedir = "${params.outdir}/pipeline_info"
+ error_strategy = 'finish'
+ allowed_error_strategies = ['terminate', 'finish', 'ignore', 'retry']
cleanup = false // if true will delete all intermediate files in work folder on workflow completion (not including staged files)
// Save of .command.* logs
@@ -85,8 +87,10 @@ params {
cleanup = params.cleanup
process {
- errorStrategy = 'finish'
+
+ errorStrategy = params.error_strategy
echo = params.echo
+
container = 'anczukowlab/splicing-pipelines-nf:3.0'
withName: 'get_accession' {
container = 'anczukowlab/download_reads:2.0'
From 558819334292f8f430c09c8762cceadb044be76a Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Tue, 28 Sep 2021 10:43:57 +0300
Subject: [PATCH 17/42] Adds Changelog to README.md
---
README.md | 35 ++++++++++++++++++++++++++++++++++-
1 file changed, 34 insertions(+), 1 deletion(-)
diff --git a/README.md b/README.md
index 5bea0911..2fcc4035 100755
--- a/README.md
+++ b/README.md
@@ -79,4 +79,37 @@ Documentation about the pipeline, found in the [`docs/`](docs) directory:
* [Running locally](docs/run_locally.md)
## Pipeline DAG
-
+
+
+
+## Changelog
+
+### v 1.1 - Pipeline improvements
+#### Fixes:
+ - Adds missing trimmomatic logs to the multiqc report
+ - Implemented correct support for input strandness in star process when `--stranded` is `second-strand` (was hardcoded to `strType=2` and only supported `first-strand` or `false` before)
+#### Updates:
+ - Updates the following tools:
+ - **STAR** `2.7.3` -> `2.7.9a` NOTE: Requires a new index! (updated in test profile)
+ - **Samtools** `1.10` -> `1.13`
+ - **StringTie** `2.1.3b` -> `2.1.7`
+ - **Gffread** `0.11.7` -> `0.12.7`
+ - multiqc `1.8` -> `1.11`
+ - deeptools `3.4.0` -> `3.5.1`
+ - bioconductor-rtracklayer `1.46.0` -> `1.52.0`
+ - gffcompare `0.11.2` -> `0.12.6`
+ - bedtools `2.29.2` -> `2.30.0`
+ - sra-tools `2.10.8` -> `2.11.0`
+ - pigz `2.3.4` -> `2.6.0`
+ - gdc-client `1.5.0` -> `1.6.1`
+ - Moves all containers to https://hub.docker.com/u/anczukowlab
+
+#### Maintenance:
+ - Consideably reduces number of basic redundant CI tests by removing completely the `max_retries` matrix and `push` from `on: [push, pull_request]`
+ - Adds CI test for sra-downloading pipeline pathway (only supported with docker profile for now)
+#### QOL:
+ - Adds saving of all the process .command* log files to results/process-logs folder
+ - Adds pipeline workdir `--cleanup` option to clear all intermediate files on pipeline successful completion
+ - Adds pipeline `--error_strategy` parameter to be able to specify pipeline error strategy directly from command line (doesn't work if specified in config linked by `-c` or `-config` nextflow params)
+
+### v 1.0 - Initial pipeline release
From b9b5fa4e6d511bddc073246d11aba0e7be3235e6 Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi
Date: Tue, 28 Sep 2021 09:40:11 +0000
Subject: [PATCH 18/42] Outsources changelog into a separate file
---
README.md | 30 +-----------------------------
changelog.md | 34 ++++++++++++++++++++++++++++++++++
2 files changed, 35 insertions(+), 29 deletions(-)
create mode 100644 changelog.md
diff --git a/README.md b/README.md
index 2fcc4035..2bc7ed04 100755
--- a/README.md
+++ b/README.md
@@ -84,32 +84,4 @@ Documentation about the pipeline, found in the [`docs/`](docs) directory:
## Changelog
-### v 1.1 - Pipeline improvements
-#### Fixes:
- - Adds missing trimmomatic logs to the multiqc report
- - Implemented correct support for input strandness in star process when `--stranded` is `second-strand` (was hardcoded to `strType=2` and only supported `first-strand` or `false` before)
-#### Updates:
- - Updates the following tools:
- - **STAR** `2.7.3` -> `2.7.9a` NOTE: Requires a new index! (updated in test profile)
- - **Samtools** `1.10` -> `1.13`
- - **StringTie** `2.1.3b` -> `2.1.7`
- - **Gffread** `0.11.7` -> `0.12.7`
- - multiqc `1.8` -> `1.11`
- - deeptools `3.4.0` -> `3.5.1`
- - bioconductor-rtracklayer `1.46.0` -> `1.52.0`
- - gffcompare `0.11.2` -> `0.12.6`
- - bedtools `2.29.2` -> `2.30.0`
- - sra-tools `2.10.8` -> `2.11.0`
- - pigz `2.3.4` -> `2.6.0`
- - gdc-client `1.5.0` -> `1.6.1`
- - Moves all containers to https://hub.docker.com/u/anczukowlab
-
-#### Maintenance:
- - Consideably reduces number of basic redundant CI tests by removing completely the `max_retries` matrix and `push` from `on: [push, pull_request]`
- - Adds CI test for sra-downloading pipeline pathway (only supported with docker profile for now)
-#### QOL:
- - Adds saving of all the process .command* log files to results/process-logs folder
- - Adds pipeline workdir `--cleanup` option to clear all intermediate files on pipeline successful completion
- - Adds pipeline `--error_strategy` parameter to be able to specify pipeline error strategy directly from command line (doesn't work if specified in config linked by `-c` or `-config` nextflow params)
-
-### v 1.0 - Initial pipeline release
+View changelog at [changelog.md](https://github.com/TheJacksonLaboratory/splicing-pipelines-nf/blob/master/changelog.md)
diff --git a/changelog.md b/changelog.md
new file mode 100644
index 00000000..0469f759
--- /dev/null
+++ b/changelog.md
@@ -0,0 +1,34 @@
+# Changelog
+
+## v 1.1 - Pipeline improvements
+
+### Fixes:
+ - Added missing trimmomatic logs to the multiqc report
+ - Implemented correct support for input strandness in star process when `--stranded` is `second-strand` (was hardcoded to `strType=2` and only supported `first-strand` or `false` before)
+
+### Updates:
+ - Updated the following tools:
+ - **STAR** `2.7.3` -> `2.7.9a` NOTE: Requires a new index! (updated in test profile)
+ - **Samtools** `1.10` -> `1.13`
+ - **StringTie** `2.1.3b` -> `2.1.7`
+ - **Gffread** `0.11.7` -> `0.12.7`
+ - multiqc `1.8` -> `1.11`
+ - deeptools `3.4.0` -> `3.5.1`
+ - bioconductor-rtracklayer `1.46.0` -> `1.52.0`
+ - gffcompare `0.11.2` -> `0.12.6`
+ - bedtools `2.29.2` -> `2.30.0`
+ - sra-tools `2.10.8` -> `2.11.0`
+ - pigz `2.3.4` -> `2.6.0`
+ - gdc-client `1.5.0` -> `1.6.1`
+ - Moved all containers to https://hub.docker.com/u/anczukowlab
+
+### Maintenance:
+ - Consideably reduced number of basic redundant CI tests by removing completely the `max_retries` matrix and `push` from `on: [push, pull_request]`
+ - Added CI test for sra-downloading pipeline pathway (only supported with docker profile for now)
+
+### Enhancements:
+ - Added saving of all the process .command* log files to results/process-logs folder
+ - Added pipeline workdir `--cleanup` option to clear all intermediate files on pipeline successful completion
+ - Added pipeline `--error_strategy` parameter to be able to specify pipeline error strategy directly from command line (doesn't work if specified in config linked by `-c` or `-config` nextflow params)
+
+## v 1.0 - Initial pipeline release
\ No newline at end of file
From ab9427a84d04fed017f1784d2694bfde5d5e1054 Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Thu, 14 Oct 2021 15:23:23 +0300
Subject: [PATCH 19/42] Fixes containers and parametrizes options in
google.config (#281)
* Fixes containers being overwritten by google.config
* Parametrize google options
---
conf/executors/google.config | 17 ++++++-----------
nextflow.config | 4 ++++
2 files changed, 10 insertions(+), 11 deletions(-)
diff --git a/conf/executors/google.config b/conf/executors/google.config
index 9aba45c5..0ff5ebda 100755
--- a/conf/executors/google.config
+++ b/conf/executors/google.config
@@ -1,16 +1,13 @@
google {
lifeSciences.bootDiskSize = params.gls_boot_disk_size
- lifeSciences.preemptible = true
- zone = 'us-east1-b'
- network = 'jax-poc-lifebit-01-vpc-network'
- subnetwork = 'us-east1-sub2'
+ lifeSciences.preemptible = params.gls_preemptible
+ zone = params.zone
+ network = params.network
+ subnetwork = params.subnetwork
}
docker.enabled = true
-executor {
- name = 'google-lifesciences'
-}
params {
@@ -21,12 +18,13 @@ params {
// disk-space allocations for stringtie_merge and rmats
// this default size is based on 100 samples
gc_disk_size = "2000 GB"
+
+ executor = 'google-lifesciences'
}
process {
maxRetries = params.max_retries
errorStrategy = { task.attempt == process.maxRetries ? 'ignore' : task.exitStatus in [3,9,10,14,143,137,104,134,139] ? 'retry' : 'ignore' }
- container = 'gcr.io/nextflow-250616/splicing-pipelines-nf:gawk'
withName: 'get_accession' {
disk = "50 GB"
cpus = {check_max(8 * task.attempt, 'cpus')}
@@ -72,13 +70,11 @@ process {
disk = params.gc_disk_size
cpus = {check_max(8 * task.attempt, 'cpus')}
memory = {check_max(30.GB * task.attempt, 'memory')}
- container = 'gcr.io/nextflow-250616/splicing-pipelines-nf:gawk'
}
withName: 'prep_de' {
disk = params.gc_disk_size
cpus = {check_max(8 * task.attempt, 'cpus')}
memory = {check_max(30.GB * task.attempt, 'memory')}
- container = 'gcr.io/nextflow-250616/splicing-pipelines-nf:gawk'
}
withName: 'rmats' {
disk = params.gc_disk_size
@@ -90,7 +86,6 @@ process {
disk = params.gc_disk_size
cpus = {check_max(30 * task.attempt, 'cpus')}
memory = {check_max(120.GB * task.attempt, 'memory')}
- container = 'gcr.io/nextflow-250616/rmats:4.1.0'
}
withName: 'multiqc' {
disk = "10 GB"
diff --git a/nextflow.config b/nextflow.config
index d2e4c2ae..c2204a2a 100755
--- a/nextflow.config
+++ b/nextflow.config
@@ -77,6 +77,10 @@ params {
// google life science specific
debug = false
gls_boot_disk_size = 50.GB
+ gls_preemptible = true
+ zone = 'us-east1-b'
+ network = 'jax-poc-lifebit-01-vpc-network'
+ subnetwork = 'us-east1-sub2'
// process scope options
echo = false
From fe7bd55a8524c10391b24ee5735961185aebd6f9 Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Thu, 28 Oct 2021 09:47:56 +0300
Subject: [PATCH 20/42] [DEL 3039] Implement ftp download for SRA accessions
(#2) (#283)
* [DEL 3039] Implement ftp download for SRA accessions (#2)
* add option for read download through FTP
* fix ftp path
* update information on download_from param
* Update run_on_sumner.md
* Fix FTP link generation; add test configs for both pair and single end data
* add catch for when single end run ends in _1
* Update main.nf
Co-authored-by: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
* Changes http to ftp in get_ftp_accession
Because this works now!!
* Make sra example data smaller
* Re-enable sra_test for singularity, now with ftp
Co-authored-by: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Co-authored-by: Vlad-Dembrovskyi
* Update docs/run_on_sumner.md
Co-authored-by: cgpu <38183826+cgpu@users.noreply.github.com>
Co-authored-by: imendes93 <73831087+imendes93@users.noreply.github.com>
Co-authored-by: cgpu <38183826+cgpu@users.noreply.github.com>
---
.github/workflows/ci.yml | 2 +-
conf/examples/sra_test.config | 2 +-
conf/examples/sra_test_paired.config | 31 ++++++++
conf/examples/sra_test_single.config | 31 ++++++++
docs/run_on_cloudos.md | 2 +-
docs/run_on_sumner.md | 2 +-
docs/usage.md | 5 +-
examples/testdata/sra/sra_test_paired_end.csv | 4 +
examples/testdata/sra/sra_test_single_end.csv | 4 +
main.nf | 76 +++++++++++++++++--
nextflow.config | 2 +
11 files changed, 148 insertions(+), 13 deletions(-)
create mode 100644 conf/examples/sra_test_paired.config
create mode 100644 conf/examples/sra_test_single.config
create mode 100644 examples/testdata/sra/sra_test_paired_end.csv
create mode 100644 examples/testdata/sra/sra_test_single_end.csv
diff --git a/.github/workflows/ci.yml b/.github/workflows/ci.yml
index ff33a593..c40899b5 100755
--- a/.github/workflows/ci.yml
+++ b/.github/workflows/ci.yml
@@ -28,7 +28,7 @@ jobs:
matrix:
singularity_version: ['3.6.4']
nxf_ver: ['20.01.0', '']
- test_type: ['ultra_quick_test']
+ test_type: ['ultra_quick_test', 'sra_test']
steps:
- uses: actions/checkout@v1
- uses: eWaterCycle/setup-singularity@v6
diff --git a/conf/examples/sra_test.config b/conf/examples/sra_test.config
index b4cb9e64..d518deae 100755
--- a/conf/examples/sra_test.config
+++ b/conf/examples/sra_test.config
@@ -12,7 +12,7 @@ params {
// Input data
singleEnd = true
reads = "$baseDir/examples/testdata/sra/sra.csv"
- download_from = 'SRA'
+ download_from = 'FTP'
// Genome references
gtf = 'https://lifebit-featured-datasets.s3-eu-west-1.amazonaws.com/projects/jax/splicing-pipelines-nf/genes.gtf'
diff --git a/conf/examples/sra_test_paired.config b/conf/examples/sra_test_paired.config
new file mode 100644
index 00000000..1d02a281
--- /dev/null
+++ b/conf/examples/sra_test_paired.config
@@ -0,0 +1,31 @@
+/*
+ * -------------------------------------------------
+ * Nextflow config file for running ultra quick tests
+ * -------------------------------------------------
+ * Defines bundled input files and everything required
+ * to run a fast and simple test. Use as follows:
+ * nextflow run jacksonlabs/splicing-pipelines-nf -profile ultra_quick_test
+ *
+ */
+
+params {
+ // Input data
+ singleEnd = false
+ reads = "$baseDir/examples/testdata/sra/sra_test_paired_end.csv"
+ download_from = 'FTP'
+
+ // Genome references
+ gtf = 'https://lifebit-featured-datasets.s3-eu-west-1.amazonaws.com/projects/jax/splicing-pipelines-nf/genes.gtf'
+ star_index = 'https://lifebit-featured-datasets.s3-eu-west-1.amazonaws.com/projects/jax/splicing-pipelines-nf/star_2.7.9a_yeast_chr_I.tar.gz'
+
+ // Other
+ test = true
+ readlength = 500
+ // This doesn't make biological sense but prevents all reads being removed during trimming
+ overhang = 100
+
+ // Limit resources
+ max_cpus = 2
+ max_memory = 6.GB
+ max_time = 48.h
+}
diff --git a/conf/examples/sra_test_single.config b/conf/examples/sra_test_single.config
new file mode 100644
index 00000000..af2422b6
--- /dev/null
+++ b/conf/examples/sra_test_single.config
@@ -0,0 +1,31 @@
+/*
+ * -------------------------------------------------
+ * Nextflow config file for running ultra quick tests
+ * -------------------------------------------------
+ * Defines bundled input files and everything required
+ * to run a fast and simple test. Use as follows:
+ * nextflow run jacksonlabs/splicing-pipelines-nf -profile ultra_quick_test
+ *
+ */
+
+params {
+ // Input data
+ singleEnd = true
+ reads = "$baseDir/examples/testdata/sra/sra_test_single_end.csv"
+ download_from = 'FTP'
+
+ // Genome references
+ gtf = 'https://lifebit-featured-datasets.s3-eu-west-1.amazonaws.com/projects/jax/splicing-pipelines-nf/genes.gtf'
+ star_index = 'https://lifebit-featured-datasets.s3-eu-west-1.amazonaws.com/projects/jax/splicing-pipelines-nf/star_2.7.9a_yeast_chr_I.tar.gz'
+
+ // Other
+ test = true
+ readlength = 500
+ // This doesn't make biological sense but prevents all reads being removed during trimming
+ overhang = 100
+
+ // Limit resources
+ max_cpus = 2
+ max_memory = 6.GB
+ max_time = 48.h
+}
diff --git a/docs/run_on_cloudos.md b/docs/run_on_cloudos.md
index 9f5a03c3..4b11adb6 100755
--- a/docs/run_on_cloudos.md
+++ b/docs/run_on_cloudos.md
@@ -29,7 +29,7 @@ If you want to start a new analysis, select `new analysis` in upper right corner
Enter all parameters as shown below. [LU BA add note about which parameter is different between cloud and sumner]. There are defaults on CloudOS just like on Sumner, but it is often good to specify each parameter you want.
-If analyzing TCGA, GTEX, or SRA, you will need to specify the `download_from` parameter. Each of these three inputs have slightly different processes that are run. For example, TCGA will download the bams and perform bamtofastq step. [For more information](https://github.com/TheJacksonLaboratory/splicing-pipelines-nf)
+If analyzing TCGA, GTEX, SRA (SRA Toolkit) or FTP (SRA FTP), you will need to specify the `download_from` parameter by either passing `TCGA`, `GTEX`, `SRA` or `FTP` respectively. Each of these inputs have slightly different processes that are run. For example, TCGA will download the bams and perform bamtofastq step. [For more information](https://github.com/TheJacksonLaboratory/splicing-pipelines-nf)
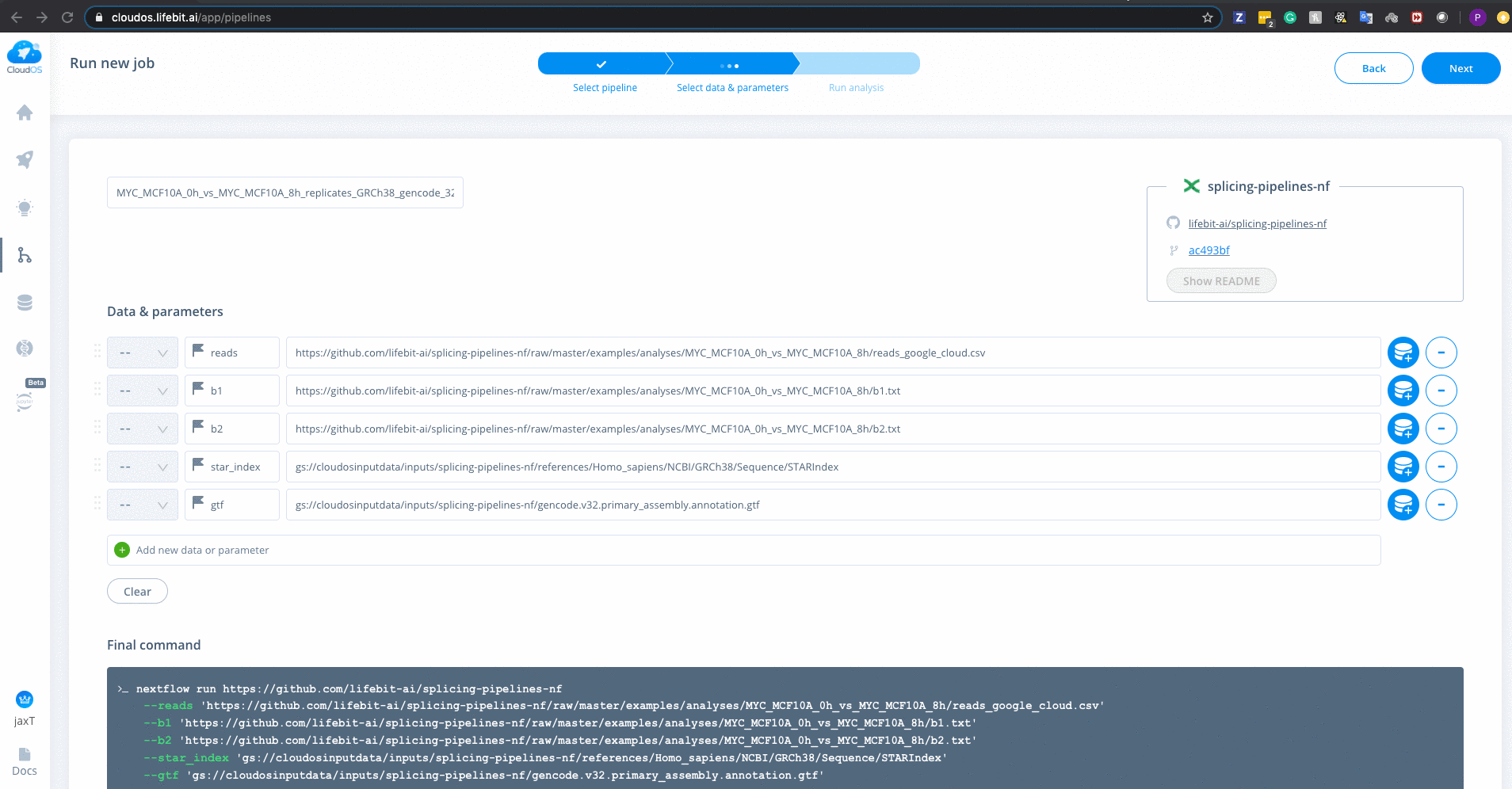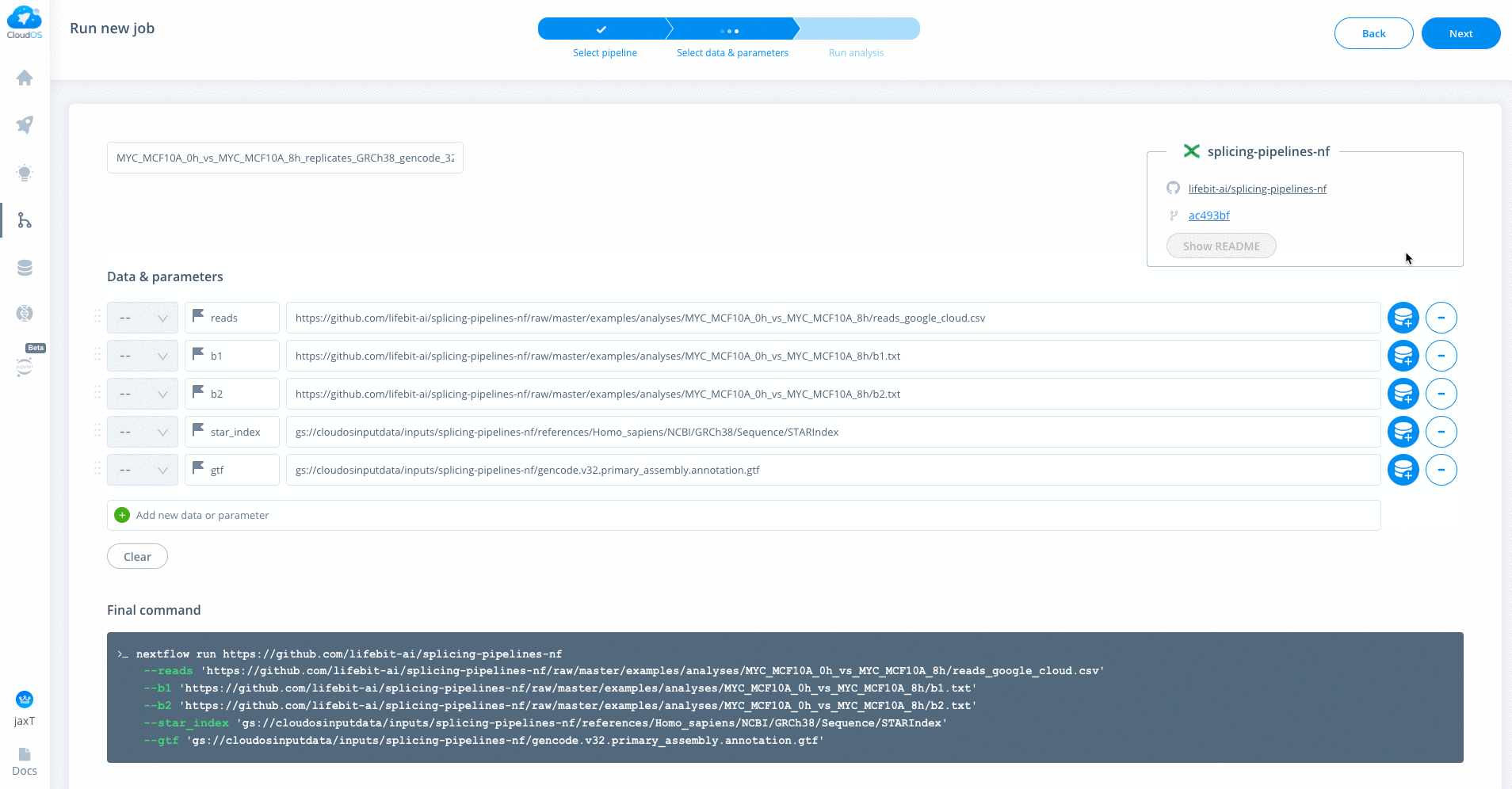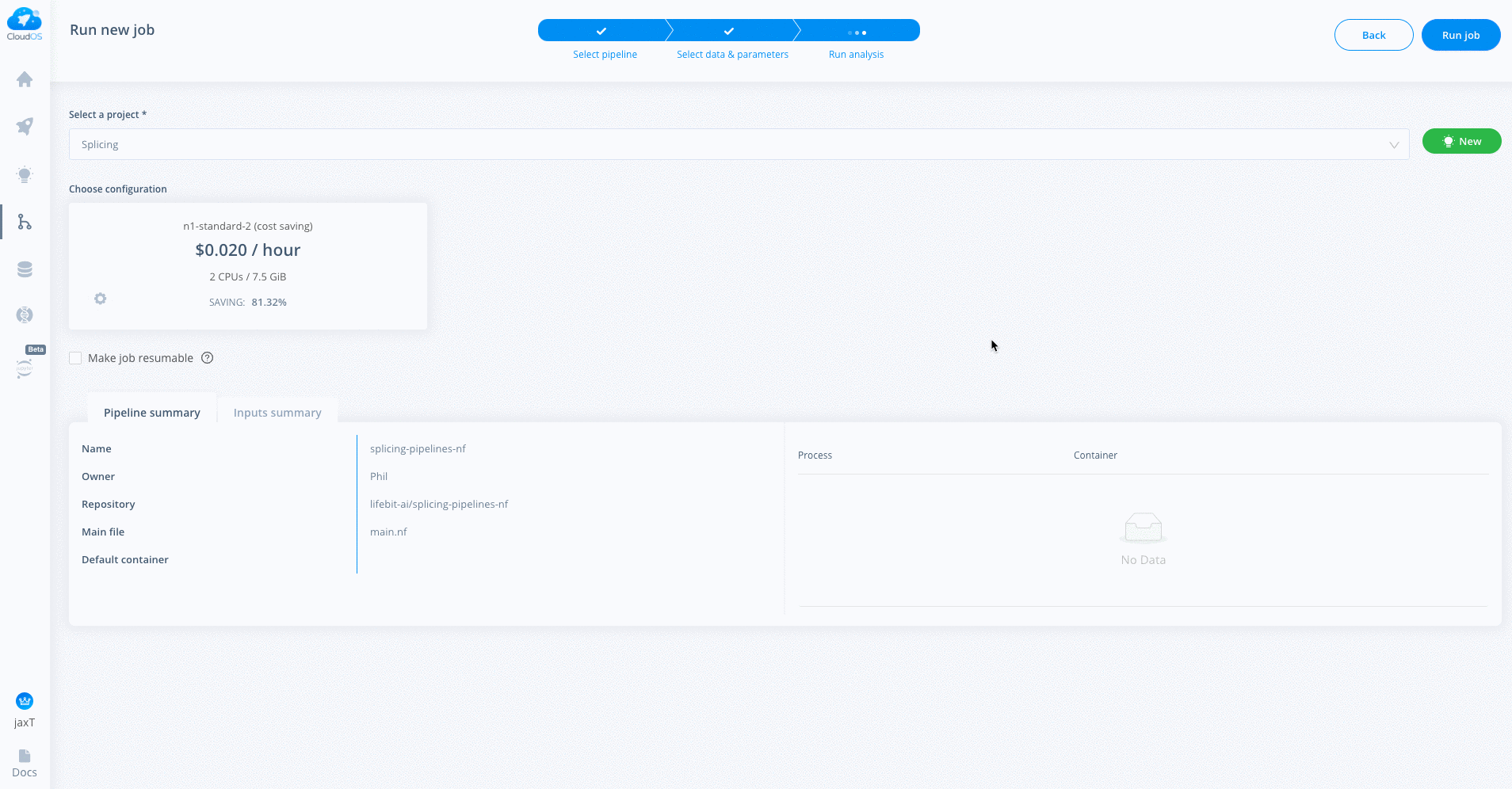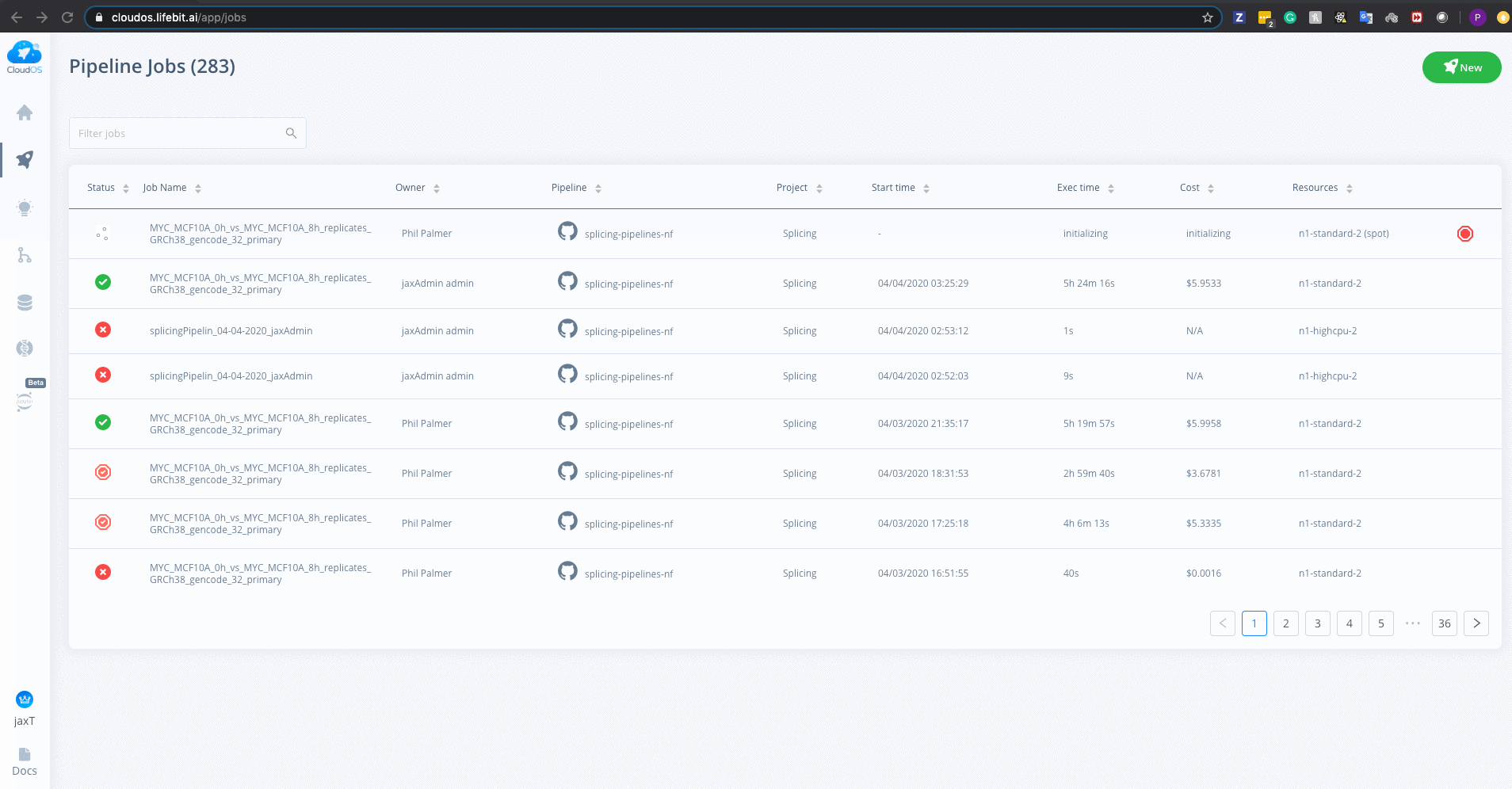
diff --git a/docs/run_on_sumner.md b/docs/run_on_sumner.md
index 22716abd..2b42f2b0 100755
--- a/docs/run_on_sumner.md
+++ b/docs/run_on_sumner.md
@@ -35,7 +35,7 @@ Input reads are specified by the `reads` input parameter, specifying a path to a
- [single-end](../examples/testdata/single_end/test_reps.csv) - must contain columns for `sample_id` and `fastq`
- [paired-end](../examples/human_test/human_test_reps.csv) - must contain columns for `sample_id`, `fastq1` and `fastq2`
-The 'reads.csv' column names must match the above [single-end] and [paired-end] examples. The `sample_id` can be anything, however each must be unique. The `fastq` column(s) should contain the path to FASTQ files. You can create this on your local computer in excel and use WinSCP to move it to Sumner, or use create it using `nano` on the cluster.
+The 'reads.csv' column names must match the above [single-end] and [paired-end] examples. The `sample_id` can be anything, however each must be unique. The `fastq` column(s) should contain the path to FASTQ files (publicly accessible ftp, s3 and gs links are also accepted). You can create this on your local computer in excel and use WinSCP to move it to Sumner, or use create it using `nano` on the cluster.
There should be one `reads.csv` file per dataset. If your dataset already has a `reads.csv` file, proceed to step 2.
diff --git a/docs/usage.md b/docs/usage.md
index dad6be73..f2ffb552 100755
--- a/docs/usage.md
+++ b/docs/usage.md
@@ -89,7 +89,8 @@ Both of these should be specified without quotes
```
Input files:
--reads Path to reads.csv file, which specifies the sample_id and path to FASTQ files for each read or read pair (path).
- This file is used if starting at beginning of pipeline.
+ This file is used if starting at beginning of pipeline. It can be file paths,
+ s3 links or ftp link.
(default: no reads.csv)
--bams Path to bams.csv file which specifies sample_id and path to BAM and BAM.bai files (path)
This file is used if starting pipeline at Stringtie.
@@ -98,7 +99,7 @@ Input files:
(default: no rmats_pairs specified)
--run_name User specified name used as prefix for output files
(defaut: no prefix, only date and time)
- --download_from Database to download FASTQ/BAMs from (available = 'TCGA', 'GTEX' or 'GEN3-DRS', 'SRA') (string)
+ --download_from Database to download FASTQ/BAMs from (available = 'TCGA', 'GTEX' or 'GEN3-DRS', 'SRA', 'FTP') (string)
false should be used to run local files on the HPC (Sumner).
'TCGA' can also be used to download GDC data including HCMI data.
(default: false)
diff --git a/examples/testdata/sra/sra_test_paired_end.csv b/examples/testdata/sra/sra_test_paired_end.csv
new file mode 100644
index 00000000..7c54df79
--- /dev/null
+++ b/examples/testdata/sra/sra_test_paired_end.csv
@@ -0,0 +1,4 @@
+sample_id
+SRR16351977
+SRR800793
+ERR6460312
\ No newline at end of file
diff --git a/examples/testdata/sra/sra_test_single_end.csv b/examples/testdata/sra/sra_test_single_end.csv
new file mode 100644
index 00000000..eaaee01b
--- /dev/null
+++ b/examples/testdata/sra/sra_test_single_end.csv
@@ -0,0 +1,4 @@
+sample_id
+ERR4667735
+SRR800793
+SRR14748080
\ No newline at end of file
diff --git a/main.nf b/main.nf
index fff032c0..33a0ef26 100755
--- a/main.nf
+++ b/main.nf
@@ -24,7 +24,8 @@ def helpMessage() {
Input files:
--reads Path to reads.csv file, which specifies the sample_id and path to FASTQ files for each read or read pair (path).
- This file is used if starting at beginning of pipeline.
+ This file is used if starting at beginning of pipeline. It can be file paths,
+ s3 links or ftp link.
(default: no reads.csv)
--bams Path to bams.csv file which specifies sample_id and path to BAM and BAM.bai files (path)
This file is used if starting pipeline at Stringtie.
@@ -33,7 +34,7 @@ def helpMessage() {
(default: no rmats_pairs specified)
--run_name User specified name used as prefix for output files
(defaut: no prefix, only date and time)
- --download_from Database to download FASTQ/BAMs from (available = 'TCGA', 'GTEX' or 'GEN3-DRS', 'SRA') (string)
+ --download_from Database to download FASTQ/BAMs from (available = 'TCGA', 'GTEX' or 'GEN3-DRS', 'SRA', 'FTP') (string)
(default: false)
--key_file For downloading reads, use TCGA authentication token (TCGA) or dbGAP repository key (GTEx, path)
or credentials.josn file in case of 'GEN3-DRS'
@@ -227,22 +228,30 @@ log.info "\n"
---------------------------------------------------*/
if (params.download_from) {
- if(download_from('gtex') || download_from('sra') || download_from('tcga')) {
+ if(download_from('gtex') || download_from('sra') || download_from('tcga') ){
Channel
.fromPath(params.reads)
.ifEmpty { exit 1, "Cannot find CSV reads file : ${params.reads}" }
.splitCsv(skip:1)
.map { sample -> sample[0].trim() }
.set { accession_ids }
- }
- if(download_from('gen3-drs')){
- Channel
+ }
+ if(download_from('gen3-drs')){
+ Channel
.fromPath(params.reads)
.ifEmpty { exit 1, "Cannot find CSV reads file : ${params.reads}" }
.splitCsv(skip:1)
.map { subj_id, file_name, md5sum, obj_id, file_size -> [subj_id, file_name, md5sum, obj_id, file_size] }
.set { ch_gtex_gen3_ids }
- }
+ }
+ if(download_from('ftp')){
+ Channel
+ .fromPath(params.reads)
+ .ifEmpty { exit 1, "Cannot find CSV reads file : ${params.reads}" }
+ .splitCsv(skip:1)
+ .map { sample -> sample[0].trim() }
+ .set { accession_ids_ftp }
+ }
}
// TODO: combine single and paired-end channel definitions
if (!params.download_from && params.singleEnd && !params.bams) {
@@ -356,6 +365,59 @@ if ( download_from('gtex') || download_from('sra') ) {
${params.savescript}
"""
}
+}
+
+if ( download_from('ftp') ) {
+ process get_ftp_accession {
+ publishDir "${params.outdir}/process-logs/${task.process}/${accession}/", pattern: "command-logs-*", mode: 'copy'
+
+ tag "${accession}"
+ label 'tiny_memory'
+
+ input:
+ val(accession) from accession_ids_ftp
+
+ output:
+ set val(accession), file(output_filename), val(params.singleEnd) into raw_reads_fastqc, raw_reads_trimmomatic
+
+ script:
+ output_filename = params.singleEnd ? "${accession}.fastq.gz" : "${accession}_{1,2}.fastq.gz"
+ isSingle = params.singleEnd ? "true" : "false"
+
+ """
+ PREFIX="\$(echo "$accession" | head -c 6)"
+ FTP_PATH="ftp://ftp.sra.ebi.ac.uk/vol1/fastq/\${PREFIX}"
+ SAMPLE=$accession
+
+ if [[ "\${#SAMPLE}" == "9" ]]; then
+ FTP_PATH="\${FTP_PATH}/$accession/$accession"
+ elif [[ "\${#SAMPLE}" == "10" ]]; then
+ SUFFIX="\${SAMPLE: -1}"
+ FTP_PATH="\${FTP_PATH}/00\${SUFFIX}/$accession/$accession"
+ elif [[ "\${#SAMPLE}" == "11" ]]; then
+ SUFFIX="\${SAMPLE: -2}"
+ FTP_PATH="\${FTP_PATH}/0\${SUFFIX}/$accession/$accession"
+ else
+ SUFFIX="\${SAMPLE: -3}"
+ FTP_PATH="\${FTP_PATH}/\${SUFFIX}/$accession/$accession"
+ fi
+
+ echo \$SAMPLE
+ echo \$FTP_PATH
+
+ if [ "$isSingle" = true ] ; then
+ {
+ wget "\${FTP_PATH}.fastq.gz"
+ } || {
+ wget "\${FTP_PATH}_1.fastq.gz"
+ mv ${accession}_1.fastq.gz ${accession}.fastq.gz
+ }
+ else
+ wget "\${FTP_PATH}_1.fastq.gz"
+ wget "\${FTP_PATH}_2.fastq.gz"
+ fi
+ """
+ }
}
/*--------------------------------------------------
diff --git a/nextflow.config b/nextflow.config
index c2204a2a..5531647f 100755
--- a/nextflow.config
+++ b/nextflow.config
@@ -138,6 +138,8 @@ profiles {
MYC_MCF10A_0h_vs_MYC_MCF10A_8h { includeConfig 'conf/examples/MYC_MCF10A_0h_vs_MYC_MCF10A_8h.config' }
ultra_quick_test { includeConfig 'conf/examples/ultra_quick_test.config' }
sra_test { includeConfig 'conf/examples/sra_test.config' }
+ sra_test_paired { includeConfig 'conf/examples/sra_test_paired.config' }
+ sra_test_single { includeConfig 'conf/examples/sra_test_single.config' }
}
dag {
From 4869322655911b6eae8cc91960b244fa0a302b35 Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Tue, 2 Nov 2021 16:31:04 -0400
Subject: [PATCH 21/42] Create Copying_Files_From_Sumner_to_Cloud
These are instructions on how to copy files from the JAX HPC Sumner to the cloud. Addresses #139
---
docs/Copying_Files_From_Sumner_to_Cloud | 34 +++++++++++++++++++++++++
1 file changed, 34 insertions(+)
create mode 100644 docs/Copying_Files_From_Sumner_to_Cloud
diff --git a/docs/Copying_Files_From_Sumner_to_Cloud b/docs/Copying_Files_From_Sumner_to_Cloud
new file mode 100644
index 00000000..80504652
--- /dev/null
+++ b/docs/Copying_Files_From_Sumner_to_Cloud
@@ -0,0 +1,34 @@
+##add singularity to $PATH:
+module load singularity
+
+## make some convenience commands to reduce typing (note we changed container name so we can accommodate other cloud providers):
+alias gcloud="singularity exec /projects/researchit/crf/containers/gcp_sdk.sif gcloud"
+alias gsutil="singularity exec /projects/researchit/crf/containers/gcp_sdk.sif gsutil"
+
+## login to gcloud; this will return a url that you need to paste into a browser, which
+## will take you through the google authentication process; you can use your jax
+## email as userid and jax password to get in. Once you authenticate, it will display
+## a code that you need to paste into the prompt provided in your ssh session on Sumner:
+
+gcloud auth login --no-launch-browser
+
+## see which projects you have access to:
+gcloud projects list
+
+## what is the project you are currently associated with:
+gcloud config list project
+
+## change project association:
+gcloud config set project my-project
+
+## see what buckets are associated with my-project:
+gsutil ls
+
+## see contents of a particular bucket:
+gsutil ls -l gs://my-bucket
+
+## recursively copy large directory from filesystem accessible on Sumner to your bucket:
+gsutil -m -o GSUtil:parallel_composite_upload_threshold=150M cp -r my_dir gs://my_bucket/my_dir
+
+## recursively copy a directory from your bucket to an existing directory on Sumner:
+gsutil -m -o GSUtil:parallel_composite_upload_threshold=150M cp -r gs://my_bucket/my_dir my_dir
From f8207567372082bb31c00c8e3e230fbddcbed67f Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Tue, 2 Nov 2021 16:31:47 -0400
Subject: [PATCH 22/42] Rename Copying_Files_From_Sumner_to_Cloud to
Copying_Files_From_Sumner_to_Cloud.md
---
...From_Sumner_to_Cloud => Copying_Files_From_Sumner_to_Cloud.md} | 0
1 file changed, 0 insertions(+), 0 deletions(-)
rename docs/{Copying_Files_From_Sumner_to_Cloud => Copying_Files_From_Sumner_to_Cloud.md} (100%)
diff --git a/docs/Copying_Files_From_Sumner_to_Cloud b/docs/Copying_Files_From_Sumner_to_Cloud.md
similarity index 100%
rename from docs/Copying_Files_From_Sumner_to_Cloud
rename to docs/Copying_Files_From_Sumner_to_Cloud.md
From 485e17b483467920c99ce4d11a5c6354a3c66052 Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Thu, 4 Nov 2021 16:41:19 +0100
Subject: [PATCH 23/42] Makes saving of unmapped files optional, cleanup true
by default (#284)
* [DEL 3039] Implement ftp download for SRA accessions (#2)
* add option for read download through FTP
* fix ftp path
* update information on download_from param
* Update run_on_sumner.md
* Fix FTP link generation; add test configs for both pair and single end data
* add catch for when single end run ends in _1
* Update main.nf
Co-authored-by: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
* Changes http to ftp in get_ftp_accession
Because this works now!!
* Make sra example data smaller
* Re-enable sra_test for singularity, now with ftp
Co-authored-by: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Co-authored-by: Vlad-Dembrovskyi
* Update docs/run_on_sumner.md
Co-authored-by: cgpu <38183826+cgpu@users.noreply.github.com>
* Add #274 , Set cleanup default as true (#3)
Co-authored-by: imendes93 <73831087+imendes93@users.noreply.github.com>
Co-authored-by: cgpu <38183826+cgpu@users.noreply.github.com>
---
main.nf | 11 ++++++++---
nextflow.config | 3 ++-
2 files changed, 10 insertions(+), 4 deletions(-)
diff --git a/main.nf b/main.nf
index 33a0ef26..9a97b48a 100755
--- a/main.nf
+++ b/main.nf
@@ -79,6 +79,8 @@ def helpMessage() {
(default: 3)
--soft_clipping Enables soft clipping (bool)
(default: true)
+ --save_unmapped Save unmapped and partially mapped reads in separate file (bool)
+ (default: false)
--star_memory Max memory to be used by STAR to sort BAM files.
(default: Available task memory)
@@ -127,7 +129,7 @@ def helpMessage() {
If pipeline is completed with errors or interrupted cleanup will not be executed. Following successfull run
resumed from the failed run with --cleanup option enabled will only clear folders of processess created in
the latest run, it will not clear cached folders coming from previous pipleine runs.
- (default: false)
+ (default: true)
See here for more info: https://github.com/TheJacksonLaboratory/splicing-pipelines-nf/blob/master/docs/usage.md
@@ -191,6 +193,7 @@ log.info "STAR index : ${star_index}"
log.info "Stranded : ${params.stranded}"
if (params.stranded) {log.info "strType : ${params.strType[params.stranded].strType}"}
log.info "Soft_clipping : ${params.soft_clipping}"
+log.info "Save unmapped : ${params.save_unmapped}"
log.info "rMATS pairs file : ${params.rmats_pairs ? params.rmats_pairs : 'Not provided'}"
log.info "Adapter : ${download_from('tcga') ? 'Will be set for each sample based based on whether the sample is paired or single-end' : adapter_file}"
log.info "Read Length : ${params.readlength}"
@@ -701,7 +704,8 @@ if (!params.bams){
tag "$name"
label 'mega_memory'
publishDir "${params.outdir}/process-logs/${task.process}/${name}", pattern: "command-logs-*", mode: 'copy'
- publishDir "${params.outdir}/star_mapped/${name}", pattern: "*{out.bam,out.bam.bai,out,ReadsPerGene.out.tab,SJ.out.tab,Unmapped}*" , mode: 'copy'
+ publishDir "${params.outdir}/star_mapped/${name}", pattern: "*{out.bam,out.bam.bai,out,ReadsPerGene.out.tab,SJ.out.tab}*" , mode: 'copy'
+ publishDir "${params.outdir}/star_mapped/${name}", pattern: "*Unmapped*", mode: 'copy'
publishDir "${params.outdir}/star_mapped/", mode: 'copy',
saveAs: {filename ->
if (filename.indexOf(".bw") > 0) "all_bigwig/${name}.bw"
@@ -732,6 +736,7 @@ if (!params.bams){
// Set maximum available memory to be used by STAR to sort BAM files
star_mem = params.star_memory ? params.star_memory : task.memory
avail_mem_bam_sort = star_mem ? "--limitBAMsortRAM ${star_mem.toBytes() - 2000000000}" : ''
+ save_unmapped_reads = params.save_unmapped ? '--outReadsUnmapped Fastx' : ''
"""
${pre_script_run_resource_status}
@@ -763,7 +768,7 @@ if (!params.bams){
--twopassMode Basic \
--alignEndsType $endsType \
--alignIntronMax 1000000 \
- --outReadsUnmapped Fastx \
+ $save_unmapped_reads \
--quantMode GeneCounts \
--outWigType None $out_filter_intron_motifs $out_sam_strand_field
diff --git a/nextflow.config b/nextflow.config
index 5531647f..166e8bae 100755
--- a/nextflow.config
+++ b/nextflow.config
@@ -43,6 +43,7 @@ params {
sjdbOverhangMin = 3
star_memory = false
soft_clipping = true
+ save_unmapped = false
// rMATS
statoff = false
@@ -62,7 +63,7 @@ params {
tracedir = "${params.outdir}/pipeline_info"
error_strategy = 'finish'
allowed_error_strategies = ['terminate', 'finish', 'ignore', 'retry']
- cleanup = false // if true will delete all intermediate files in work folder on workflow completion (not including staged files)
+ cleanup = true // if true will delete all intermediate files in work folder on workflow completion (not including staged files)
// Save of .command.* logs
savescript = 'task_hash=`basename \${PWD} | cut -c1-6`; mkdir command-logs-\$task_hash ; cp .command.*{err,log,sh} command-logs-\$task_hash'
From 089d6e3ded62d85d05a0cf213691cac533836a8b Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi
Date: Thu, 4 Nov 2021 16:47:48 +0000
Subject: [PATCH 24/42] Sets default --cleanup false for google.config
---
conf/executors/google.config | 2 ++
1 file changed, 2 insertions(+)
diff --git a/conf/executors/google.config b/conf/executors/google.config
index 0ff5ebda..4200f406 100755
--- a/conf/executors/google.config
+++ b/conf/executors/google.config
@@ -19,6 +19,8 @@ params {
// this default size is based on 100 samples
gc_disk_size = "2000 GB"
+ cleanup = false // Don't change, otherwise CloudOS jobs won't be resumable by default even if user wants to.
+
executor = 'google-lifesciences'
}
From 3b71e038b186bb2bc92debacb02aede7b5dae917 Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Tue, 9 Nov 2021 09:58:12 +0200
Subject: [PATCH 25/42] Adds --save_unmapped to usage.md
---
docs/usage.md | 2 ++
1 file changed, 2 insertions(+)
diff --git a/docs/usage.md b/docs/usage.md
index f2ffb552..8caea353 100755
--- a/docs/usage.md
+++ b/docs/usage.md
@@ -150,6 +150,8 @@ Star:
If false, the STAR parameter will be --alignEndsType 'EndToEnd' and no rMATS parameter is added.
NOTE: Soft Clipping will cause read lengths to be variable, so turn soft_clipping off if reads need to be same length. Variable read length parameter is turned on in rMATS when minlen does not equal readlength.
(default: true)
+ --save_unmapped Save unmapped and partially mapped reads in separate file (bool)
+ (default: false)
--star_memory Max memory to be used by STAR to sort BAM files.
(default: Available task memory)
From 0745c1f8fbdc5f6c2fbe4e4ed051f58111a143ba Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Wed, 10 Nov 2021 17:10:21 -0500
Subject: [PATCH 26/42] Update usage.md
Updating config info
---
docs/usage.md | 22 ++++++++++++++++------
1 file changed, 16 insertions(+), 6 deletions(-)
diff --git a/docs/usage.md b/docs/usage.md
index 8caea353..3c8060c6 100755
--- a/docs/usage.md
+++ b/docs/usage.md
@@ -93,7 +93,7 @@ Input files:
s3 links or ftp link.
(default: no reads.csv)
--bams Path to bams.csv file which specifies sample_id and path to BAM and BAM.bai files (path)
- This file is used if starting pipeline at Stringtie.
+ If this file is provided, pipeline will start at Stringtie (and proceed through rMATS and post processing).
(default: no bams.csv)
--rmats_pairs Path to rmats_pairs.txt file containing b1 (and b2) samples names (path)
(default: no rmats_pairs specified)
@@ -113,8 +113,11 @@ Main arguments:
--assembly_name Genome assembly name (available = 'GRCh38' or 'GRCm38', string)
(default: false)
--star_index Path to STAR index (path)
- (default: read length)
+ Star indices must be generated prior to run (with correct STAR version)
+ (default: false)
--singleEnd Specifies that the input is single-end reads (bool)
+ This parameter also automatically establishes the path to the SE or PE adapters.
+ For PE, set to false.
(default: false)
--stranded Specifies that the input is stranded ('first-strand', 'second-strand', false (aka unstranded))
(default: 'first-strand')
@@ -122,6 +125,8 @@ Main arguments:
--readlength Read length - Note that all reads will be cropped to this length(int)
(default: no read length specified)
-profile Configuration profile to use. Can use multiple (comma separated, string)
+ On sumner, this should be set in the main.pbs or as a command-line parameter.
+ Profile can only be activated from the command line.
Available: base, docker, sumner, test and more.
Trimmomatic:
@@ -142,11 +147,13 @@ Star:
--overhang Overhang (int)
(default: readlength - 1)
--filterScore Controls --outFilterScoreMinOverLread and outFilterMatchNminOverLread
+ For TCGA values: https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
(default: 0.66)
--sjdbOverhangMin Controls --alignSJDBoverhangMin (int)
+ For TCGA values: https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
(default: 3)
--soft_clipping Enables soft clipping (bool)
- If true, the STAR parameter will be --alignEndsType Local and the rMATS parameter --allow-clipping will be added.
+ If true, the STAR parameter will be --alignEndsType 'Local' and the rMATS parameter --allow-clipping will be added.
If false, the STAR parameter will be --alignEndsType 'EndToEnd' and no rMATS parameter is added.
NOTE: Soft Clipping will cause read lengths to be variable, so turn soft_clipping off if reads need to be same length. Variable read length parameter is turned on in rMATS when minlen does not equal readlength.
(default: true)
@@ -170,6 +177,7 @@ rMATS:
Other:
--test For running trim test (bool)
+ To run the first half of the pipeline (through STAR), set test = true.
(default: false)
--max_cpus Maximum number of CPUs (int)
(default: 72)
@@ -182,7 +190,7 @@ Other:
--skipMultiQC Skip MultiQC (bool)
(default: false)
--outdir The output directory where the results will be saved (string)
- On Sumner, this must be set in the main.pbs. NF_splicing_pipeline.config will not overwrite main.pbs.
+ On Sumner, this must be set in the main.pbs or via command line. NF_splicing_pipeline.config will not overwrite main.pbs.
(default: directory where you submit the job)
--mega_time Sets time limit for processes withLabel 'mega_memory' in the main.nf using the base.config (time unit)
Make sure '#SBATCH -t' in 'main.pbs' is appropriately set if you are changing this parameter.
@@ -195,16 +203,18 @@ Other:
--error_strategy Mode of pipeline handling failed processes. Possible values: 'terminate', 'finish', 'ignore', 'retry'.
Check nextflow documnetation for detailed descriptions of each mode:
https://www.nextflow.io/docs/latest/process.html#process-page-error-strategy
- (default: finish)
+ Set this parameter in the main.pbs, on the command line, or see NF_splicing_pipeline.config example (does not work like normal config param)
+ This does not overwrited CloudOS config (set to 'errorStrategy = { task.exitStatus in [3,9,10,14,143,137,104,134,139] ? 'retry': 'ignore'}
+ (default: 'finish')
--cleanup This option will enable nextflow work folder cleanup upon pipeline successfull completion.
All intermediate files from nexftlow processes' workdirs will be cleared, staging folder with staged
files will not be cleared.
If pipeline is completed with errors or interrupted cleanup will not be executed. Following successfull run
resumed from the failed run with --cleanup option enabled will only clear folders of processess created in
the latest run, it will not clear cached folders coming from previous pipleine runs.
+ Set this parameter in the main.pbs, on the command line, or see NF_splicing_pipeline.config example (does not work like normal config param)
(default: false)
-
```
From 6ff6a5d3a629b2a9923f99c8266f7120409f4371 Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Wed, 10 Nov 2021 17:14:48 -0500
Subject: [PATCH 27/42] Create NF_splicing_pipeline.config
Example NF Config
---
NF_splicing_pipeline.config | 52 +++++++++++++++++++++++++++++++++++++
1 file changed, 52 insertions(+)
create mode 100644 NF_splicing_pipeline.config
diff --git a/NF_splicing_pipeline.config b/NF_splicing_pipeline.config
new file mode 100644
index 00000000..0410f31b
--- /dev/null
+++ b/NF_splicing_pipeline.config
@@ -0,0 +1,52 @@
+params {
+ // Input data:
+ reads = 'reads.csv'
+ rmats_pairs = 'rmats_pairs.txt'
+ run_name = 'B6_finalrun'
+ download_from = false
+ key_file = false
+
+ // Main arguments:
+ gtf = '/projects/anczukow-lab/reference_genomes/mouse_black6/Gencode/gencode.vM23.primary_assembly.annotation.gtf'
+ assembly_name = 'GRCm38'
+ star_index = '/projects/anczukow-lab/reference_genomes/mouse_black6/Gencode/star_overhangs_2.7.9a/star_2.7.9a_GRCm38_150.tar.gz'
+ singleEnd = false
+ stranded = 'first-strand'
+ readlength = 150
+
+ // Trimmomatic:
+ minlen = 20
+ slidingwindow = true
+
+ //Star:
+ mismatch = 5
+ filterScore = 0.66
+ sjdbOverhangMin = 3
+ soft_clipping = true
+ save_unmapped = false
+
+ //rMATS:
+ statoff = false
+ paired_stats = false
+ novelSS = false
+ mil = 50
+ mel = 500
+
+ //Other:
+ test = false
+ max_cpus = 72
+ max_memory = 760.GB
+ max_time = 72.h
+ skiprMATS = false
+ skipMultiQC = false
+ mega_time = 20.h
+ debug = false
+ error_strategy = 'finish'
+ cleanup = false
+}
+
+cleanup = params.cleanup
+process {
+ errorStrategy = params.error_strategy
+ ...
+}
From 9fa6719e812ad452ac4a6f898ffa051db5fc83ed Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Wed, 10 Nov 2021 17:17:12 -0500
Subject: [PATCH 28/42] Update run_on_sumner.md
---
docs/run_on_sumner.md | 2 +-
1 file changed, 1 insertion(+), 1 deletion(-)
diff --git a/docs/run_on_sumner.md b/docs/run_on_sumner.md
index 2b42f2b0..731d5ac8 100755
--- a/docs/run_on_sumner.md
+++ b/docs/run_on_sumner.md
@@ -76,7 +76,7 @@ Each rMATS comparison must be specified with a comparison name as well as the `s
This config file will be specific to your user and analysis. **You do not need to edit the pipeline code to configure the pipeline**. Descriptions of all possible parameters and their default values can be found [here](usage.md#all-available-parameters).
-To create your own custom config (to specify your input parameters) you can copy and edit this [example config](../conf/examples/MYC_MCF10A_0h_vs_MYC_MCF10A_8h.config) file.
+To create your own custom config (to specify your input parameters) you can copy and edit this [example config](NF_splicing_pipeline.config) file.
**VERY IMPORTANT NOTES***
From 847e463b536ee467fbc91b02f1a40729acd4b2f1 Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Wed, 10 Nov 2021 17:17:46 -0500
Subject: [PATCH 29/42] Update run_on_sumner.md
---
docs/run_on_sumner.md | 2 +-
1 file changed, 1 insertion(+), 1 deletion(-)
diff --git a/docs/run_on_sumner.md b/docs/run_on_sumner.md
index 731d5ac8..20361771 100755
--- a/docs/run_on_sumner.md
+++ b/docs/run_on_sumner.md
@@ -76,7 +76,7 @@ Each rMATS comparison must be specified with a comparison name as well as the `s
This config file will be specific to your user and analysis. **You do not need to edit the pipeline code to configure the pipeline**. Descriptions of all possible parameters and their default values can be found [here](usage.md#all-available-parameters).
-To create your own custom config (to specify your input parameters) you can copy and edit this [example config](NF_splicing_pipeline.config) file.
+To create your own custom config (to specify your input parameters) you can copy and edit this [example config](https://github.com/TheJacksonLaboratory/splicing-pipelines-nf/blob/6ff6a5d3a629b2a9923f99c8266f7120409f4371/NF_splicing_pipeline.config) file.
**VERY IMPORTANT NOTES***
From 26453265cb316c26e8c0e96f56a5d4b1513b98b3 Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Wed, 10 Nov 2021 17:40:35 -0500
Subject: [PATCH 30/42] Update usage.md
---
docs/usage.md | 8 ++++----
1 file changed, 4 insertions(+), 4 deletions(-)
diff --git a/docs/usage.md b/docs/usage.md
index 3c8060c6..110d93f3 100755
--- a/docs/usage.md
+++ b/docs/usage.md
@@ -104,7 +104,7 @@ Input files:
'TCGA' can also be used to download GDC data including HCMI data.
(default: false)
--key_file For downloading reads, use TCGA authentication token (TCGA) or dbGAP repository key (GTEx, path)
- or credentials.josn file in case of 'GEN3-DRS'
+ or credentials.json file in case of 'GEN3-DRS'
(default: false)
Main arguments:
@@ -120,8 +120,8 @@ Main arguments:
For PE, set to false.
(default: false)
--stranded Specifies that the input is stranded ('first-strand', 'second-strand', false (aka unstranded))
- (default: 'first-strand')
- 'first-strand' refers to RF/fr-firststrand in this pipeline.
+ 'first-strand' refers to RF/fr-firststrand in this pipeline.
+ (default: 'first-strand')
--readlength Read length - Note that all reads will be cropped to this length(int)
(default: no read length specified)
-profile Configuration profile to use. Can use multiple (comma separated, string)
@@ -182,7 +182,7 @@ Other:
--max_cpus Maximum number of CPUs (int)
(default: 72)
--max_memory Maximum memory (memory unit)
- (default: 760)
+ (default: 760.GB)
--max_time Maximum time (time unit)
(default: 72.h)
--skiprMATS Skip rMATS (bool)
From 1a73179e8f3023673dd90d7f68f40071293662ca Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Wed, 10 Nov 2021 17:41:17 -0500
Subject: [PATCH 31/42] Update main.nf
Updating to match usage.md parameter descriptions
---
main.nf | 37 ++++++++++++++++++++++++++++---------
1 file changed, 28 insertions(+), 9 deletions(-)
diff --git a/main.nf b/main.nf
index 9a97b48a..1572919c 100755
--- a/main.nf
+++ b/main.nf
@@ -28,13 +28,15 @@ def helpMessage() {
s3 links or ftp link.
(default: no reads.csv)
--bams Path to bams.csv file which specifies sample_id and path to BAM and BAM.bai files (path)
- This file is used if starting pipeline at Stringtie.
+ If this file is provided, pipeline will start at Stringtie (and proceed through rMATS and post processing).
(default: no bams.csv)
--rmats_pairs Path to rmats_pairs.txt file containing b1 (and b2) samples names (path)
(default: no rmats_pairs specified)
--run_name User specified name used as prefix for output files
(defaut: no prefix, only date and time)
--download_from Database to download FASTQ/BAMs from (available = 'TCGA', 'GTEX' or 'GEN3-DRS', 'SRA', 'FTP') (string)
+ false should be used to run local files on the HPC (Sumner).
+ 'TCGA' can also be used to download GDC data including HCMI data.
(default: false)
--key_file For downloading reads, use TCGA authentication token (TCGA) or dbGAP repository key (GTEx, path)
or credentials.josn file in case of 'GEN3-DRS'
@@ -46,20 +48,26 @@ def helpMessage() {
--assembly_name Genome assembly name (available = 'GRCh38' or 'GRCm38', string)
(default: false)
--star_index Path to STAR index (path)
- (default: read length)
+ Star indices must be generated prior to run (with correct STAR version)
+ (default: false)
--singleEnd Specifies that the input is single-end reads (bool)
+ This parameter also automatically establishes the path to the SE or PE adapters.
+ For PE, set to false.
(default: false)
--stranded Specifies that the input is stranded ('first-strand', 'second-strand', false (aka unstranded))
+ 'first-strand' refers to RF/fr-firststrand in this pipeline.
(default: 'first-strand')
--readlength Read length - Note that all reads will be cropped to this length(int)
(default: no read length specified)
-profile Configuration profile to use. Can use multiple (comma separated, string)
+ On sumner, this should be set in the main.pbs or as a command-line parameter.
+ Profile can only be activated from the command line.
Available: base, docker, sumner, test and more.
Trimmomatic:
--minlen Drop the read if it is below a specified length (int)
Default parameters turn on --variable-readlength
- To crop all reads and turn off, set minlen = readlength
+ To crop all reads and turn off --variable-readlength, set minlen = readlength
(default: 20)
--slidingwindow Perform a sliding window trimming approach (bool)
(default: true)
@@ -74,10 +82,15 @@ def helpMessage() {
--overhang Overhang (int)
(default: readlength - 1)
--filterScore Controls --outFilterScoreMinOverLread and outFilterMatchNminOverLread
+ For TCGA values: https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
(default: 0.66)
--sjdbOverhangMin Controls --alignSJDBoverhangMin (int)
+ For TCGA values: https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
(default: 3)
--soft_clipping Enables soft clipping (bool)
+ If true, the STAR parameter will be --alignEndsType 'Local' and the rMATS parameter --allow-clipping will be added.
+ If false, the STAR parameter will be --alignEndsType 'EndToEnd' and no rMATS parameter is added.
+ NOTE: Soft Clipping will cause read lengths to be variable, so turn soft_clipping off if reads need to be same length. Variable read length parameter is turned on in rMATS when minlen does not equal readlength.
(default: true)
--save_unmapped Save unmapped and partially mapped reads in separate file (bool)
(default: false)
@@ -99,20 +112,23 @@ def helpMessage() {
Other:
--test For running trim test (bool)
+ To run the first half of the pipeline (through STAR), set test = true.
(default: false)
--max_cpus Maximum number of CPUs (int)
- (default: ?)
+ (default: 72)
--max_memory Maximum memory (memory unit)
- (default: 80)
+ (default: 760.GB)
--max_time Maximum time (time unit)
- (default: ?)
+ (default: 72.h)
--skiprMATS Skip rMATS (bool)
(default: false)
--skipMultiQC Skip MultiQC (bool)
(default: false)
--outdir The output directory where the results will be saved (string)
+ On Sumner, this must be set in the main.pbs or via command line. NF_splicing_pipeline.config will not overwrite main.pbs.
(default: directory where you submit the job)
- --mega_time Sets time limit for processes withLabel 'mega_memory' in the main.nf using the base.config (time unit)
+ --mega_time Sets time limit for processes withLabel 'mega_memory' in the main.nf using the base.config (time unit)
+ Make sure '#SBATCH -t' in 'main.pbs' is appropriately set if you are changing this parameter.
(default: 20.h)
--gc_disk_size Only specific to google-cloud executor. Adds disk-space for few aggregative processes.
(default: "200 GB" based on 100 samples. Simply add 2 x Number of Samples)
@@ -122,13 +138,16 @@ def helpMessage() {
--error_strategy Mode of pipeline handling failed processes. Possible values: 'terminate', 'finish', 'ignore', 'retry'.
Check nextflow documnetation for detailed descriptions of each mode:
https://www.nextflow.io/docs/latest/process.html#process-page-error-strategy
- (default: $params.error_strategy)
+ Set this parameter in the main.pbs, on the command line, or see NF_splicing_pipeline.config example (does not work like normal config param)
+ This does not overwrited CloudOS config (set to 'errorStrategy = { task.exitStatus in [3,9,10,14,143,137,104,134,139] ? 'retry': 'ignore'}
+ (default: 'finish')
--cleanup This option will enable nextflow work folder cleanup upon pipeline successfull completion.
All intermediate files from nexftlow processes' workdirs will be cleared, staging folder with staged
files will not be cleared.
If pipeline is completed with errors or interrupted cleanup will not be executed. Following successfull run
resumed from the failed run with --cleanup option enabled will only clear folders of processess created in
the latest run, it will not clear cached folders coming from previous pipleine runs.
+ Set this parameter in the main.pbs, on the command line, or see NF_splicing_pipeline.config example (does not work like normal config param)
(default: true)
@@ -1173,4 +1192,4 @@ workflow.onComplete {
log.info "-${c_purple}[splicing-pipelines-nf]${c_red} Cleanup: Working directory was not cleared from intermediate files due to pipeline errors. You can re-use them with -resume option. ${c_reset}-"
}
}
-}
\ No newline at end of file
+}
From 77c96c4d3f5fe39a13a6a27b8e632913c355506c Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Wed, 10 Nov 2021 17:48:48 -0500
Subject: [PATCH 32/42] Update main.nf
---
main.nf | 2 +-
1 file changed, 1 insertion(+), 1 deletion(-)
diff --git a/main.nf b/main.nf
index 1572919c..b8b5dc4d 100755
--- a/main.nf
+++ b/main.nf
@@ -148,7 +148,7 @@ def helpMessage() {
resumed from the failed run with --cleanup option enabled will only clear folders of processess created in
the latest run, it will not clear cached folders coming from previous pipleine runs.
Set this parameter in the main.pbs, on the command line, or see NF_splicing_pipeline.config example (does not work like normal config param)
- (default: true)
+ (default: config.process.errorStrategy)
See here for more info: https://github.com/TheJacksonLaboratory/splicing-pipelines-nf/blob/master/docs/usage.md
From 03c977a4a5b386a1ea31c8aae78592432e38f3a2 Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Thu, 11 Nov 2021 18:25:45 +0200
Subject: [PATCH 33/42] set errorStrategy to finish in base.config for sumner
---
conf/executors/base.config | 2 +-
1 file changed, 1 insertion(+), 1 deletion(-)
diff --git a/conf/executors/base.config b/conf/executors/base.config
index 14f9234f..ccfcb895 100755
--- a/conf/executors/base.config
+++ b/conf/executors/base.config
@@ -15,7 +15,7 @@ params {
process {
- errorStrategy = { task.exitStatus in [143,137,104,134,139] ? 'retry' : 'terminate' }
+ errorStrategy = 'finish'
maxRetries = params.max_retries
maxErrors = '-1'
From 9739c03e5c0306ae65735615ffd990bc5ce8c619 Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Thu, 11 Nov 2021 11:57:48 -0500
Subject: [PATCH 34/42] Update run_on_sumner.md
---
docs/run_on_sumner.md | 6 ++++--
1 file changed, 4 insertions(+), 2 deletions(-)
diff --git a/docs/run_on_sumner.md b/docs/run_on_sumner.md
index 20361771..4b49ded1 100755
--- a/docs/run_on_sumner.md
+++ b/docs/run_on_sumner.md
@@ -74,7 +74,7 @@ Each rMATS comparison must be specified with a comparison name as well as the `s
### 4. Setup `NF_splicing_pipeline.config`
-This config file will be specific to your user and analysis. **You do not need to edit the pipeline code to configure the pipeline**. Descriptions of all possible parameters and their default values can be found [here](usage.md#all-available-parameters).
+This config file will be specific to your user and analysis. **You do not need to edit the pipeline code to configure the pipeline**. Descriptions of all possible parameters and their default values can be found [here](usage.md#all-available-parameters) and [here](https://github.com/TheJacksonLaboratory/splicing-pipelines-nf/blob/03c977a4a5b386a1ea31c8aae78592432e38f3a2/nextflow.config).
To create your own custom config (to specify your input parameters) you can copy and edit this [example config](https://github.com/TheJacksonLaboratory/splicing-pipelines-nf/blob/6ff6a5d3a629b2a9923f99c8266f7120409f4371/NF_splicing_pipeline.config) file.
@@ -152,7 +152,9 @@ Each rMATS comparison must be specified with a comparison name as well as the `s
### 4. Setup `NF_splicing_pipeline.config`
-This config file will be specific to your user and analysis. **You do not need to edit the pipeline code to configure the pipeline**. Descriptions of all possible parameters and their default values can be found [here](usage.md#all-available-parameters).
+This config file will be specific to your user and analysis. **You do not need to edit the pipeline code to configure the pipeline**. Descriptions of all possible parameters and their default values can be found [here](usage.md#all-available-parameters) and [here](https://github.com/TheJacksonLaboratory/splicing-pipelines-nf/blob/03c977a4a5b386a1ea31c8aae78592432e38f3a2/nextflow.config).
+
+To create your own custom config (to specify your input parameters) you can copy and edit this [example config]() file.
**VERY IMPORTANT NOTES***
From 9b9d854a2969af821ec009f3bb28293bfc21226b Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Thu, 11 Nov 2021 19:44:03 +0200
Subject: [PATCH 35/42] Update help message
---
main.nf | 108 +++++++++++++++++++++++++++++++++-----------------------
1 file changed, 63 insertions(+), 45 deletions(-)
diff --git a/main.nf b/main.nf
index b8b5dc4d..63f004f2 100755
--- a/main.nf
+++ b/main.nf
@@ -21,30 +21,34 @@ def helpMessage() {
Usage:
The typical command for running the pipeline is as follows:
nextflow run main.nf --reads my_reads.csv --gtf genome.gtf --star_index star_dir -profile base,sumner
-
+
Input files:
- --reads Path to reads.csv file, which specifies the sample_id and path to FASTQ files for each read or read pair (path).
+ --reads Path to reads.csv file, which specifies the sample_id and path to FASTQ files
+ for each read or read pair (path).
This file is used if starting at beginning of pipeline. It can be file paths,
s3 links or ftp link.
(default: no reads.csv)
- --bams Path to bams.csv file which specifies sample_id and path to BAM and BAM.bai files (path)
- If this file is provided, pipeline will start at Stringtie (and proceed through rMATS and post processing).
+ --bams Path to bams.csv file which specifies sample_id and path to BAM and BAM.bai
+ files (path)
+ If this file is provided, pipeline will start at Stringtie (and proceed through
+ rMATS and post processing).
(default: no bams.csv)
--rmats_pairs Path to rmats_pairs.txt file containing b1 (and b2) samples names (path)
- (default: no rmats_pairs specified)
+ (default: no rmats_pairs specified)
--run_name User specified name used as prefix for output files
(defaut: no prefix, only date and time)
- --download_from Database to download FASTQ/BAMs from (available = 'TCGA', 'GTEX' or 'GEN3-DRS', 'SRA', 'FTP') (string)
+ --download_from Database to download FASTQ/BAMs from (available = 'TCGA', 'GTEX' or 'GEN3-DRS',
+ 'SRA', 'FTP') (string)
false should be used to run local files on the HPC (Sumner).
- 'TCGA' can also be used to download GDC data including HCMI data.
+ 'TCGA' can also be used to download GDC data including HCMI data.
+ (default: false)
+ --key_file For downloading reads, use TCGA authentication token (TCGA) or dbGAP repository
+ key (GTEx, path) or credentials.json file in case of 'GEN3-DRS'
(default: false)
- --key_file For downloading reads, use TCGA authentication token (TCGA) or dbGAP repository key (GTEx, path)
- or credentials.josn file in case of 'GEN3-DRS'
- (default: false)
-
+
Main arguments:
--gtf Path to reference GTF file (path)
- (default: no gtf specified)
+ (default: no gtf specified)
--assembly_name Genome assembly name (available = 'GRCh38' or 'GRCm38', string)
(default: false)
--star_index Path to STAR index (path)
@@ -52,52 +56,59 @@ def helpMessage() {
(default: false)
--singleEnd Specifies that the input is single-end reads (bool)
This parameter also automatically establishes the path to the SE or PE adapters.
- For PE, set to false.
+ For PE, set to false.
(default: false)
- --stranded Specifies that the input is stranded ('first-strand', 'second-strand', false (aka unstranded))
+ --stranded Specifies that the input is stranded ('first-strand', 'second-strand',
+ false (aka unstranded))
'first-strand' refers to RF/fr-firststrand in this pipeline.
(default: 'first-strand')
--readlength Read length - Note that all reads will be cropped to this length(int)
- (default: no read length specified)
+ (default: no read length specified)
-profile Configuration profile to use. Can use multiple (comma separated, string)
On sumner, this should be set in the main.pbs or as a command-line parameter.
- Profile can only be activated from the command line.
+ Profile can only be activated from the command line.
Available: base, docker, sumner, test and more.
- Trimmomatic:
+ Trimmomatic:
--minlen Drop the read if it is below a specified length (int)
Default parameters turn on --variable-readlength
- To crop all reads and turn off --variable-readlength, set minlen = readlength
+ To crop all reads and turn off --variable-readlength, set minlen = readlength
(default: 20)
--slidingwindow Perform a sliding window trimming approach (bool)
(default: true)
- --adapter Path to adapter file (path)
+ --adapter Path to adapter file (path)
(default: TruSeq3 for either PE or SE, see singleEnd parameter)
-
- Star:
+
+ Star:
--mismatch Number of allowed mismatches per read (SE) or combined read (PE) (int)
SE ex. read length of 50, allow 2 mismatches per 50 bp
- PE ex. read length of 50, allow 2 mismatches per 100 bp
+ PE ex. read length of 50, allow 2 mismatches per 100 bp
(default: 2)
--overhang Overhang (int)
(default: readlength - 1)
--filterScore Controls --outFilterScoreMinOverLread and outFilterMatchNminOverLread
- For TCGA values: https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
+ For TCGA values:
+ https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
(default: 0.66)
--sjdbOverhangMin Controls --alignSJDBoverhangMin (int)
- For TCGA values: https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
+ For TCGA values:
+ https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
(default: 3)
--soft_clipping Enables soft clipping (bool)
- If true, the STAR parameter will be --alignEndsType 'Local' and the rMATS parameter --allow-clipping will be added.
- If false, the STAR parameter will be --alignEndsType 'EndToEnd' and no rMATS parameter is added.
- NOTE: Soft Clipping will cause read lengths to be variable, so turn soft_clipping off if reads need to be same length. Variable read length parameter is turned on in rMATS when minlen does not equal readlength.
+ If true, the STAR parameter will be --alignEndsType 'Local' and the rMATS parameter
+ --allow-clipping will be added.
+ If false, the STAR parameter will be --alignEndsType 'EndToEnd' and no rMATS
+ parameter is added.
+ NOTE: Soft Clipping will cause read lengths to be variable, so turn soft_clipping
+ off if reads need to be same length. Variable read length parameter is turned on
+ in rMATS when minlen does not equal readlength.
(default: true)
--save_unmapped Save unmapped and partially mapped reads in separate file (bool)
(default: false)
--star_memory Max memory to be used by STAR to sort BAM files.
(default: Available task memory)
- rMATS:
+ rMATS:
--statoff Skip the statistical analysis (bool)
If using only b1 as input, this must be turned on.
(default: false)
@@ -115,7 +126,7 @@ def helpMessage() {
To run the first half of the pipeline (through STAR), set test = true.
(default: false)
--max_cpus Maximum number of CPUs (int)
- (default: 72)
+ (default: 72)
--max_memory Maximum memory (memory unit)
(default: 760.GB)
--max_time Maximum time (time unit)
@@ -125,30 +136,37 @@ def helpMessage() {
--skipMultiQC Skip MultiQC (bool)
(default: false)
--outdir The output directory where the results will be saved (string)
- On Sumner, this must be set in the main.pbs or via command line. NF_splicing_pipeline.config will not overwrite main.pbs.
- (default: directory where you submit the job)
- --mega_time Sets time limit for processes withLabel 'mega_memory' in the main.nf using the base.config (time unit)
+ On Sumner, this must be set in the main.pbs or via command line.
+ NF_splicing_pipeline.config will not overwrite main.pbs.
+ (default: /results)
+ --mega_time Sets time limit for processes withLabel 'mega_memory' in the main.nf using the
+ base.config (time unit)
Make sure '#SBATCH -t' in 'main.pbs' is appropriately set if you are changing this parameter.
(default: 20.h)
--gc_disk_size Only specific to google-cloud executor. Adds disk-space for few aggregative processes.
(default: "200 GB" based on 100 samples. Simply add 2 x Number of Samples)
- --debug This option will enable echo of script execution into STDOUT with some additional
+ --debug This option will enable echo of script execution into STDOUT with some additional
resource information (such as machine type, memory, cpu and disk space)
(default: false)
- --error_strategy Mode of pipeline handling failed processes. Possible values: 'terminate', 'finish', 'ignore', 'retry'.
+ --error_strategy Mode of pipeline handling failed processes.
+ Possible values: 'terminate', 'finish', 'ignore', 'retry'.
Check nextflow documnetation for detailed descriptions of each mode:
https://www.nextflow.io/docs/latest/process.html#process-page-error-strategy
- Set this parameter in the main.pbs, on the command line, or see NF_splicing_pipeline.config example (does not work like normal config param)
- This does not overwrited CloudOS config (set to 'errorStrategy = { task.exitStatus in [3,9,10,14,143,137,104,134,139] ? 'retry': 'ignore'}
- (default: 'finish')
- --cleanup This option will enable nextflow work folder cleanup upon pipeline successfull completion.
- All intermediate files from nexftlow processes' workdirs will be cleared, staging folder with staged
- files will not be cleared.
- If pipeline is completed with errors or interrupted cleanup will not be executed. Following successfull run
- resumed from the failed run with --cleanup option enabled will only clear folders of processess created in
- the latest run, it will not clear cached folders coming from previous pipleine runs.
- Set this parameter in the main.pbs, on the command line, or see NF_splicing_pipeline.config example (does not work like normal config param)
- (default: config.process.errorStrategy)
+ Set this parameter in the main.pbs, on the command line, or see NF_splicing_pipeline.config
+ example (does not work like normal config param)
+ This does not overwrited CloudOS config, which is set to:
+ 'errorStrategy = { task.exitStatus in [3,9,10,14,143,137,104,134,139] ? 'retry': 'ignore'}
+ (default (non-cloudos): 'finish')
+ --cleanup This option will enable nextflow work folder cleanup upon pipeline successfull
+ completion. All intermediate files from nexftlow processes' workdirs will be
+ cleared, staging folder with staged files will not be cleared.
+ If pipeline is completed with errors or interrupted cleanup will not be executed.
+ Following successfull run resumed from the failed run with --cleanup option enabled
+ will only clear folders of processess created in the latest run, it will not clear
+ cached folders coming from previous pipleine runs.
+ Set this parameter in the main.pbs, on the command line, or see NF_splicing_pipeline.config
+ example (does not work like normal config param)
+ (default non-cloudos: true; cloudos: false)
See here for more info: https://github.com/TheJacksonLaboratory/splicing-pipelines-nf/blob/master/docs/usage.md
From fae6d5bfaa2256eb18b270e2af9a4a26e7e7daed Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Thu, 11 Nov 2021 19:50:00 +0200
Subject: [PATCH 36/42] Update usage.md to match main.nf help message
---
docs/usage.md | 172 +++++++++++++++++++++++++++-----------------------
1 file changed, 94 insertions(+), 78 deletions(-)
diff --git a/docs/usage.md b/docs/usage.md
index 110d93f3..6bf8f5c7 100755
--- a/docs/usage.md
+++ b/docs/usage.md
@@ -88,134 +88,150 @@ Both of these should be specified without quotes
## All available parameters
```
Input files:
- --reads Path to reads.csv file, which specifies the sample_id and path to FASTQ files for each read or read pair (path).
+ --reads Path to reads.csv file, which specifies the sample_id and path to FASTQ files
+ for each read or read pair (path).
This file is used if starting at beginning of pipeline. It can be file paths,
s3 links or ftp link.
(default: no reads.csv)
- --bams Path to bams.csv file which specifies sample_id and path to BAM and BAM.bai files (path)
- If this file is provided, pipeline will start at Stringtie (and proceed through rMATS and post processing).
+ --bams Path to bams.csv file which specifies sample_id and path to BAM and BAM.bai
+ files (path)
+ If this file is provided, pipeline will start at Stringtie (and proceed through
+ rMATS and post processing).
(default: no bams.csv)
- --rmats_pairs Path to rmats_pairs.txt file containing b1 (and b2) samples names (path)
- (default: no rmats_pairs specified)
- --run_name User specified name used as prefix for output files
+ --rmats_pairs Path to rmats_pairs.txt file containing b1 (and b2) samples names (path)
+ (default: no rmats_pairs specified)
+ --run_name User specified name used as prefix for output files
(defaut: no prefix, only date and time)
- --download_from Database to download FASTQ/BAMs from (available = 'TCGA', 'GTEX' or 'GEN3-DRS', 'SRA', 'FTP') (string)
- false should be used to run local files on the HPC (Sumner).
- 'TCGA' can also be used to download GDC data including HCMI data.
+ --download_from Database to download FASTQ/BAMs from (available = 'TCGA', 'GTEX' or 'GEN3-DRS',
+ 'SRA', 'FTP') (string)
+ false should be used to run local files on the HPC (Sumner).
+ 'TCGA' can also be used to download GDC data including HCMI data.
(default: false)
- --key_file For downloading reads, use TCGA authentication token (TCGA) or dbGAP repository key (GTEx, path)
- or credentials.json file in case of 'GEN3-DRS'
+ --key_file For downloading reads, use TCGA authentication token (TCGA) or dbGAP repository
+ key (GTEx, path) or credentials.json file in case of 'GEN3-DRS'
(default: false)
-
+
Main arguments:
- --gtf Path to reference GTF file (path)
- (default: no gtf specified)
- --assembly_name Genome assembly name (available = 'GRCh38' or 'GRCm38', string)
+ --gtf Path to reference GTF file (path)
+ (default: no gtf specified)
+ --assembly_name Genome assembly name (available = 'GRCh38' or 'GRCm38', string)
(default: false)
- --star_index Path to STAR index (path)
- Star indices must be generated prior to run (with correct STAR version)
+ --star_index Path to STAR index (path)
+ Star indices must be generated prior to run (with correct STAR version)
(default: false)
- --singleEnd Specifies that the input is single-end reads (bool)
+ --singleEnd Specifies that the input is single-end reads (bool)
This parameter also automatically establishes the path to the SE or PE adapters.
For PE, set to false.
(default: false)
- --stranded Specifies that the input is stranded ('first-strand', 'second-strand', false (aka unstranded))
+ --stranded Specifies that the input is stranded ('first-strand', 'second-strand',
+ false (aka unstranded))
'first-strand' refers to RF/fr-firststrand in this pipeline.
- (default: 'first-strand')
- --readlength Read length - Note that all reads will be cropped to this length(int)
+ (default: 'first-strand')
+ --readlength Read length - Note that all reads will be cropped to this length(int)
(default: no read length specified)
- -profile Configuration profile to use. Can use multiple (comma separated, string)
+ -profile Configuration profile to use. Can use multiple (comma separated, string)
On sumner, this should be set in the main.pbs or as a command-line parameter.
- Profile can only be activated from the command line.
+ Profile can only be activated from the command line.
Available: base, docker, sumner, test and more.
-
-Trimmomatic:
- --minlen Drop the read if it is below a specified length (int)
- Default parameters turn on --variable-readlength
- To crop all reads and turn off --variable-readlength, set minlen = readlength
+
+Trimmomatic:
+ --minlen Drop the read if it is below a specified length (int)
+ Default parameters turn on --variable-readlength
+ To crop all reads and turn off --variable-readlength, set minlen = readlength
(default: 20)
- --slidingwindow Perform a sliding window trimming approach (bool)
+ --slidingwindow Perform a sliding window trimming approach (bool)
(default: true)
- --adapter Path to adapter file (path)
+ --adapter Path to adapter file (path)
(default: TruSeq3 for either PE or SE, see singleEnd parameter)
-
-Star:
- --mismatch Number of allowed mismatches per read (SE) or combined read (PE) (int)
+
+Star:
+ --mismatch Number of allowed mismatches per read (SE) or combined read (PE) (int)
SE ex. read length of 50, allow 2 mismatches per 50 bp
- PE ex. read length of 50, allow 2 mismatches per 100 bp
+ PE ex. read length of 50, allow 2 mismatches per 100 bp
(default: 2)
- --overhang Overhang (int)
+ --overhang Overhang (int)
(default: readlength - 1)
- --filterScore Controls --outFilterScoreMinOverLread and outFilterMatchNminOverLread
- For TCGA values: https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
+ --filterScore Controls --outFilterScoreMinOverLread and outFilterMatchNminOverLread
+ For TCGA values:
+ https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
(default: 0.66)
- --sjdbOverhangMin Controls --alignSJDBoverhangMin (int)
- For TCGA values: https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
+ --sjdbOverhangMin Controls --alignSJDBoverhangMin (int)
+ For TCGA values:
+ https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
(default: 3)
- --soft_clipping Enables soft clipping (bool)
- If true, the STAR parameter will be --alignEndsType 'Local' and the rMATS parameter --allow-clipping will be added.
- If false, the STAR parameter will be --alignEndsType 'EndToEnd' and no rMATS parameter is added.
- NOTE: Soft Clipping will cause read lengths to be variable, so turn soft_clipping off if reads need to be same length. Variable read length parameter is turned on in rMATS when minlen does not equal readlength.
+ --soft_clipping Enables soft clipping (bool)
+ If true, the STAR parameter will be --alignEndsType 'Local' and the rMATS parameter
+ --allow-clipping will be added.
+ If false, the STAR parameter will be --alignEndsType 'EndToEnd' and no rMATS
+ parameter is added.
+ NOTE: Soft Clipping will cause read lengths to be variable, so turn soft_clipping
+ off if reads need to be same length. Variable read length parameter is turned on
+ in rMATS when minlen does not equal readlength.
(default: true)
- --save_unmapped Save unmapped and partially mapped reads in separate file (bool)
+ --save_unmapped Save unmapped and partially mapped reads in separate file (bool)
(default: false)
- --star_memory Max memory to be used by STAR to sort BAM files.
+ --star_memory Max memory to be used by STAR to sort BAM files.
(default: Available task memory)
-rMATS:
- --statoff Skip the statistical analysis (bool)
+rMATS:
+ --statoff Skip the statistical analysis (bool)
If using only b1 as input, this must be turned on.
(default: false)
- --paired_stats Use the paired stats model (bool)
+ --paired_stats Use the paired stats model (bool)
(default: false)
- --novelSS Enable detection of unnanotated splice sites (bool)
+ --novelSS Enable detection of unnanotated splice sites (bool)
(default: false)
- --mil Minimum Intron Length. Only impacts --novelSS behavior (int)
+ --mil Minimum Intron Length. Only impacts --novelSS behavior (int)
(default: 50)
- --mel Maximum Exon Length. Only impacts --novelSS behavior (int)
+ --mel Maximum Exon Length. Only impacts --novelSS behavior (int)
(default: 500)
Other:
- --test For running trim test (bool)
+ --test For running trim test (bool)
To run the first half of the pipeline (through STAR), set test = true.
(default: false)
- --max_cpus Maximum number of CPUs (int)
- (default: 72)
- --max_memory Maximum memory (memory unit)
+ --max_cpus Maximum number of CPUs (int)
+ (default: 72)
+ --max_memory Maximum memory (memory unit)
(default: 760.GB)
- --max_time Maximum time (time unit)
+ --max_time Maximum time (time unit)
(default: 72.h)
- --skiprMATS Skip rMATS (bool)
+ --skiprMATS Skip rMATS (bool)
(default: false)
- --skipMultiQC Skip MultiQC (bool)
+ --skipMultiQC Skip MultiQC (bool)
(default: false)
- --outdir The output directory where the results will be saved (string)
- On Sumner, this must be set in the main.pbs or via command line. NF_splicing_pipeline.config will not overwrite main.pbs.
- (default: directory where you submit the job)
- --mega_time Sets time limit for processes withLabel 'mega_memory' in the main.nf using the base.config (time unit)
+ --outdir The output directory where the results will be saved (string)
+ On Sumner, this must be set in the main.pbs or via command line.
+ NF_splicing_pipeline.config will not overwrite main.pbs.
+ (default: /results)
+ --mega_time Sets time limit for processes withLabel 'mega_memory' in the main.nf using the
+ base.config (time unit)
Make sure '#SBATCH -t' in 'main.pbs' is appropriately set if you are changing this parameter.
(default: 20.h)
- --gc_disk_size Only specific to google-cloud executor. Adds disk-space for few aggregative processes.
+ --gc_disk_size Only specific to google-cloud executor. Adds disk-space for few aggregative processes.
(default: "200 GB" based on 100 samples. Simply add 2 x Number of Samples)
- --debug This option will enable echo of script execution into STDOUT with some additional
+ --debug This option will enable echo of script execution into STDOUT with some additional
resource information (such as machine type, memory, cpu and disk space)
(default: false)
- --error_strategy Mode of pipeline handling failed processes. Possible values: 'terminate', 'finish', 'ignore', 'retry'.
+ --error_strategy Mode of pipeline handling failed processes.
+ Possible values: 'terminate', 'finish', 'ignore', 'retry'.
Check nextflow documnetation for detailed descriptions of each mode:
https://www.nextflow.io/docs/latest/process.html#process-page-error-strategy
- Set this parameter in the main.pbs, on the command line, or see NF_splicing_pipeline.config example (does not work like normal config param)
- This does not overwrited CloudOS config (set to 'errorStrategy = { task.exitStatus in [3,9,10,14,143,137,104,134,139] ? 'retry': 'ignore'}
- (default: 'finish')
- --cleanup This option will enable nextflow work folder cleanup upon pipeline successfull completion.
- All intermediate files from nexftlow processes' workdirs will be cleared, staging folder with staged
- files will not be cleared.
- If pipeline is completed with errors or interrupted cleanup will not be executed. Following successfull run
- resumed from the failed run with --cleanup option enabled will only clear folders of processess created in
- the latest run, it will not clear cached folders coming from previous pipleine runs.
- Set this parameter in the main.pbs, on the command line, or see NF_splicing_pipeline.config example (does not work like normal config param)
- (default: false)
-
-
+ Set this parameter in the main.pbs, on the command line, or see NF_splicing_pipeline.config
+ example (does not work like normal config param)
+ This does not overwrited CloudOS config, which is set to:
+ 'errorStrategy = { task.exitStatus in [3,9,10,14,143,137,104,134,139] ? 'retry': 'ignore'}
+ (default (non-cloudos): 'finish')
+ --cleanup This option will enable nextflow work folder cleanup upon pipeline successfull
+ completion. All intermediate files from nexftlow processes' workdirs will be
+ cleared, staging folder with staged files will not be cleared.
+ If pipeline is completed with errors or interrupted cleanup will not be executed.
+ Following successfull run resumed from the failed run with --cleanup option enabled
+ will only clear folders of processess created in the latest run, it will not clear
+ cached folders coming from previous pipleine runs.
+ Set this parameter in the main.pbs, on the command line, or see NF_splicing_pipeline.config
+ example (does not work like normal config param)
+ (default non-cloudos: true; cloudos: false)
```
## Run with data from AnviL Gen3-DRS
From 560c7bef762a17fa6973c3a9a66cd224ab8b56de Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Thu, 11 Nov 2021 19:56:42 +0200
Subject: [PATCH 37/42] Update usage.md to fix indentations
---
docs/usage.md | 78 +++++++++++++++++++++++++--------------------------
1 file changed, 39 insertions(+), 39 deletions(-)
diff --git a/docs/usage.md b/docs/usage.md
index 6bf8f5c7..6e12128c 100755
--- a/docs/usage.md
+++ b/docs/usage.md
@@ -88,46 +88,46 @@ Both of these should be specified without quotes
## All available parameters
```
Input files:
- --reads Path to reads.csv file, which specifies the sample_id and path to FASTQ files
+ --reads Path to reads.csv file, which specifies the sample_id and path to FASTQ files
for each read or read pair (path).
This file is used if starting at beginning of pipeline. It can be file paths,
s3 links or ftp link.
(default: no reads.csv)
- --bams Path to bams.csv file which specifies sample_id and path to BAM and BAM.bai
+ --bams Path to bams.csv file which specifies sample_id and path to BAM and BAM.bai
files (path)
If this file is provided, pipeline will start at Stringtie (and proceed through
rMATS and post processing).
(default: no bams.csv)
- --rmats_pairs Path to rmats_pairs.txt file containing b1 (and b2) samples names (path)
+ --rmats_pairs Path to rmats_pairs.txt file containing b1 (and b2) samples names (path)
(default: no rmats_pairs specified)
- --run_name User specified name used as prefix for output files
+ --run_name User specified name used as prefix for output files
(defaut: no prefix, only date and time)
- --download_from Database to download FASTQ/BAMs from (available = 'TCGA', 'GTEX' or 'GEN3-DRS',
+ --download_from Database to download FASTQ/BAMs from (available = 'TCGA', 'GTEX' or 'GEN3-DRS',
'SRA', 'FTP') (string)
false should be used to run local files on the HPC (Sumner).
'TCGA' can also be used to download GDC data including HCMI data.
(default: false)
- --key_file For downloading reads, use TCGA authentication token (TCGA) or dbGAP repository
+ --key_file For downloading reads, use TCGA authentication token (TCGA) or dbGAP repository
key (GTEx, path) or credentials.json file in case of 'GEN3-DRS'
(default: false)
Main arguments:
- --gtf Path to reference GTF file (path)
+ --gtf Path to reference GTF file (path)
(default: no gtf specified)
- --assembly_name Genome assembly name (available = 'GRCh38' or 'GRCm38', string)
+ --assembly_name Genome assembly name (available = 'GRCh38' or 'GRCm38', string)
(default: false)
- --star_index Path to STAR index (path)
+ --star_index Path to STAR index (path)
Star indices must be generated prior to run (with correct STAR version)
(default: false)
- --singleEnd Specifies that the input is single-end reads (bool)
+ --singleEnd Specifies that the input is single-end reads (bool)
This parameter also automatically establishes the path to the SE or PE adapters.
For PE, set to false.
(default: false)
- --stranded Specifies that the input is stranded ('first-strand', 'second-strand',
+ --stranded Specifies that the input is stranded ('first-strand', 'second-strand',
false (aka unstranded))
'first-strand' refers to RF/fr-firststrand in this pipeline.
(default: 'first-strand')
- --readlength Read length - Note that all reads will be cropped to this length(int)
+ --readlength Read length - Note that all reads will be cropped to this length(int)
(default: no read length specified)
-profile Configuration profile to use. Can use multiple (comma separated, string)
On sumner, this should be set in the main.pbs or as a command-line parameter.
@@ -135,31 +135,31 @@ Main arguments:
Available: base, docker, sumner, test and more.
Trimmomatic:
- --minlen Drop the read if it is below a specified length (int)
+ --minlen Drop the read if it is below a specified length (int)
Default parameters turn on --variable-readlength
To crop all reads and turn off --variable-readlength, set minlen = readlength
(default: 20)
- --slidingwindow Perform a sliding window trimming approach (bool)
+ --slidingwindow Perform a sliding window trimming approach (bool)
(default: true)
- --adapter Path to adapter file (path)
+ --adapter Path to adapter file (path)
(default: TruSeq3 for either PE or SE, see singleEnd parameter)
Star:
- --mismatch Number of allowed mismatches per read (SE) or combined read (PE) (int)
+ --mismatch Number of allowed mismatches per read (SE) or combined read (PE) (int)
SE ex. read length of 50, allow 2 mismatches per 50 bp
PE ex. read length of 50, allow 2 mismatches per 100 bp
(default: 2)
- --overhang Overhang (int)
+ --overhang Overhang (int)
(default: readlength - 1)
- --filterScore Controls --outFilterScoreMinOverLread and outFilterMatchNminOverLread
+ --filterScore Controls --outFilterScoreMinOverLread and outFilterMatchNminOverLread
For TCGA values:
https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
(default: 0.66)
- --sjdbOverhangMin Controls --alignSJDBoverhangMin (int)
+ --sjdbOverhangMin Controls --alignSJDBoverhangMin (int)
For TCGA values:
https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/
(default: 3)
- --soft_clipping Enables soft clipping (bool)
+ --soft_clipping Enables soft clipping (bool)
If true, the STAR parameter will be --alignEndsType 'Local' and the rMATS parameter
--allow-clipping will be added.
If false, the STAR parameter will be --alignEndsType 'EndToEnd' and no rMATS
@@ -168,52 +168,52 @@ Star:
off if reads need to be same length. Variable read length parameter is turned on
in rMATS when minlen does not equal readlength.
(default: true)
- --save_unmapped Save unmapped and partially mapped reads in separate file (bool)
+ --save_unmapped Save unmapped and partially mapped reads in separate file (bool)
(default: false)
- --star_memory Max memory to be used by STAR to sort BAM files.
+ --star_memory Max memory to be used by STAR to sort BAM files.
(default: Available task memory)
rMATS:
- --statoff Skip the statistical analysis (bool)
+ --statoff Skip the statistical analysis (bool)
If using only b1 as input, this must be turned on.
(default: false)
- --paired_stats Use the paired stats model (bool)
+ --paired_stats Use the paired stats model (bool)
(default: false)
- --novelSS Enable detection of unnanotated splice sites (bool)
+ --novelSS Enable detection of unnanotated splice sites (bool)
(default: false)
- --mil Minimum Intron Length. Only impacts --novelSS behavior (int)
+ --mil Minimum Intron Length. Only impacts --novelSS behavior (int)
(default: 50)
- --mel Maximum Exon Length. Only impacts --novelSS behavior (int)
+ --mel Maximum Exon Length. Only impacts --novelSS behavior (int)
(default: 500)
Other:
- --test For running trim test (bool)
+ --test For running trim test (bool)
To run the first half of the pipeline (through STAR), set test = true.
(default: false)
- --max_cpus Maximum number of CPUs (int)
+ --max_cpus Maximum number of CPUs (int)
(default: 72)
- --max_memory Maximum memory (memory unit)
+ --max_memory Maximum memory (memory unit)
(default: 760.GB)
- --max_time Maximum time (time unit)
+ --max_time Maximum time (time unit)
(default: 72.h)
- --skiprMATS Skip rMATS (bool)
+ --skiprMATS Skip rMATS (bool)
(default: false)
- --skipMultiQC Skip MultiQC (bool)
+ --skipMultiQC Skip MultiQC (bool)
(default: false)
- --outdir The output directory where the results will be saved (string)
+ --outdir The output directory where the results will be saved (string)
On Sumner, this must be set in the main.pbs or via command line.
NF_splicing_pipeline.config will not overwrite main.pbs.
(default: /results)
- --mega_time Sets time limit for processes withLabel 'mega_memory' in the main.nf using the
+ --mega_time Sets time limit for processes withLabel 'mega_memory' in the main.nf using the
base.config (time unit)
Make sure '#SBATCH -t' in 'main.pbs' is appropriately set if you are changing this parameter.
(default: 20.h)
- --gc_disk_size Only specific to google-cloud executor. Adds disk-space for few aggregative processes.
+ --gc_disk_size Only specific to google-cloud executor. Adds disk-space for few aggregative processes.
(default: "200 GB" based on 100 samples. Simply add 2 x Number of Samples)
- --debug This option will enable echo of script execution into STDOUT with some additional
+ --debug This option will enable echo of script execution into STDOUT with some additional
resource information (such as machine type, memory, cpu and disk space)
(default: false)
- --error_strategy Mode of pipeline handling failed processes.
+ --error_strategy Mode of pipeline handling failed processes.
Possible values: 'terminate', 'finish', 'ignore', 'retry'.
Check nextflow documnetation for detailed descriptions of each mode:
https://www.nextflow.io/docs/latest/process.html#process-page-error-strategy
@@ -222,7 +222,7 @@ Other:
This does not overwrited CloudOS config, which is set to:
'errorStrategy = { task.exitStatus in [3,9,10,14,143,137,104,134,139] ? 'retry': 'ignore'}
(default (non-cloudos): 'finish')
- --cleanup This option will enable nextflow work folder cleanup upon pipeline successfull
+ --cleanup This option will enable nextflow work folder cleanup upon pipeline successfull
completion. All intermediate files from nexftlow processes' workdirs will be
cleared, staging folder with staged files will not be cleared.
If pipeline is completed with errors or interrupted cleanup will not be executed.
From 7c15665287e8eece1b88e0b8fe962b2c94fb256f Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Thu, 11 Nov 2021 13:17:15 -0500
Subject: [PATCH 38/42] Update usage.md
Moving NF tips from "running pipeline on Sumner"
---
docs/usage.md | 16 ++++++++++++++--
1 file changed, 14 insertions(+), 2 deletions(-)
diff --git a/docs/usage.md b/docs/usage.md
index 6e12128c..9c077bbd 100755
--- a/docs/usage.md
+++ b/docs/usage.md
@@ -14,6 +14,7 @@ See [here](run_locally.md)
Nextflow parameters can be provided in one of two ways:
1) They can be specified in configuration files
2) They can be specified on the command-line
+3) They can be specified in main.pbs
For example, all of the following are equivalent:
@@ -44,9 +45,9 @@ nextflow run main.nf --reads /path/to/reads.csv --readlength 48 --singleEnd true
-Parameters specified on the command-line take precedence over those specified in configuration files. It is generally best-practice to have your parameters saved in a configuration file as this makes your analysis more reproducible if you need to run it again.
+Parameters specified on the command-line (or in main.pbs) take precedence over those specified in configuration files. It is generally best-practice to have your parameters saved in a configuration file as this makes your analysis more reproducible if you need to run it again.
-[Profiles](https://www.nextflow.io/docs/latest/en/latest/config.html#config-profiles) are configuration that can be included by specifying the profile name on the command-line. For example, `-profile sumner` to include configuration specific to JAX's HPC Sumner
+[Profiles](https://www.nextflow.io/docs/latest/en/latest/config.html#config-profiles) are configuration that can be included by specifying the profile name on the command-line. This CANNOT be set from the configuration file. For example, `-profile sumner` to include configuration specific to JAX's HPC Sumner.
## Types of Nextflow parameters
@@ -234,6 +235,17 @@ Other:
(default non-cloudos: true; cloudos: false)
```
+# Useful Nextflow options
+
+Whereas parameters are set on the command-line using double dash options eg `--reads`, parameters passed to Nextflow itself can be provided with single-dash options eg `-profile`.
+
+You can see some of these options [here](https://www.nextflow.io/docs/latest/tracing.html) in the Nextflow documentation.
+
+Some useful ones include (specified in main.pbs):
+- `-resume` which will [resume](https://www.nextflow.io/docs/latest/getstarted.html?highlight=resume#modify-and-resume) any cached processes that have not been changed
+- `-with-trace` eg `-with-trace trace.txt` which gives a [trace report](https://www.nextflow.io/docs/latest/tracing.html?highlight=dag#trace-report) for resource consumption by the pipeline
+- `-with-dag` eg `-with-dag flowchart.png` which produces the [DAG visualisation](https://www.nextflow.io/docs/latest/tracing.html?highlight=dag#dag-visualisation) graph showing each of the different processes and the connections between them (the channels)
+
## Run with data from AnviL Gen3-DRS
You will be needing two things from - https://gen3.theanvil.io/
From 35e159a8d098955f6992e1532f47eebade437ef6 Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Thu, 11 Nov 2021 13:18:04 -0500
Subject: [PATCH 39/42] Update run_on_sumner.md
Moved NF tips to usage.md
---
docs/run_on_sumner.md | 11 -----------
1 file changed, 11 deletions(-)
diff --git a/docs/run_on_sumner.md b/docs/run_on_sumner.md
index 4b49ded1..3017244e 100755
--- a/docs/run_on_sumner.md
+++ b/docs/run_on_sumner.md
@@ -176,14 +176,3 @@ Run the pipeline!
```
sbatch /projects/anczukow-lab/splicing_pipeline/splicing-pipelines-nf/main.pbs
```
-
-# Bonus: useful Nextflow options
-
-Whereas parameters are set on the command-line using double dash options eg `--reads`, parameters passed to Nextflow itself can be provided with single-dash options eg `-profile`.
-
-You can see some of these options [here](https://www.nextflow.io/docs/latest/tracing.html) in the Nextflow documentation.
-
-Some useful ones include (specified in main.pbs):
-- `-resume` which will [resume](https://www.nextflow.io/docs/latest/getstarted.html?highlight=resume#modify-and-resume) any cached processes that have not been changed
-- `-with-trace` eg `-with-trace trace.txt` which gives a [trace report](https://www.nextflow.io/docs/latest/tracing.html?highlight=dag#trace-report) for resource consumption by the pipeline
-- `-with-dag` eg `-with-dag flowchart.png` which produces the [DAG visualisation](https://www.nextflow.io/docs/latest/tracing.html?highlight=dag#dag-visualisation) graph showing each of the different processes and the connections between them (the channels)
From ef65fe2ac92e22e657f1e7a7d31d35d2876f7e0e Mon Sep 17 00:00:00 2001
From: angarb <62404570+angarb@users.noreply.github.com>
Date: Wed, 17 Nov 2021 17:26:39 -0500
Subject: [PATCH 40/42] Update Copying_Files_From_Sumner_to_Cloud.md
---
docs/Copying_Files_From_Sumner_to_Cloud.md | 26 +++++++++++-----------
1 file changed, 13 insertions(+), 13 deletions(-)
diff --git a/docs/Copying_Files_From_Sumner_to_Cloud.md b/docs/Copying_Files_From_Sumner_to_Cloud.md
index 80504652..6a9f36bb 100644
--- a/docs/Copying_Files_From_Sumner_to_Cloud.md
+++ b/docs/Copying_Files_From_Sumner_to_Cloud.md
@@ -1,34 +1,34 @@
-##add singularity to $PATH:
+//add singularity to $PATH:
module load singularity
-## make some convenience commands to reduce typing (note we changed container name so we can accommodate other cloud providers):
+//make some convenience commands to reduce typing (note we changed container name so we can accommodate other cloud providers):
alias gcloud="singularity exec /projects/researchit/crf/containers/gcp_sdk.sif gcloud"
alias gsutil="singularity exec /projects/researchit/crf/containers/gcp_sdk.sif gsutil"
-## login to gcloud; this will return a url that you need to paste into a browser, which
-## will take you through the google authentication process; you can use your jax
-## email as userid and jax password to get in. Once you authenticate, it will display
-## a code that you need to paste into the prompt provided in your ssh session on Sumner:
+//login to gcloud; this will return a url that you need to paste into a browser, which
+//will take you through the google authentication process; you can use your jax
+//email as userid and jax password to get in. Once you authenticate, it will display
+//a code that you need to paste into the prompt provided in your ssh session on Sumner:
gcloud auth login --no-launch-browser
-## see which projects you have access to:
+//see which projects you have access to:
gcloud projects list
-## what is the project you are currently associated with:
+//what is the project you are currently associated with:
gcloud config list project
-## change project association:
+//change project association:
gcloud config set project my-project
-## see what buckets are associated with my-project:
+//see what buckets are associated with my-project:
gsutil ls
-## see contents of a particular bucket:
+//see contents of a particular bucket:
gsutil ls -l gs://my-bucket
-## recursively copy large directory from filesystem accessible on Sumner to your bucket:
+//recursively copy large directory from filesystem accessible on Sumner to your bucket:
gsutil -m -o GSUtil:parallel_composite_upload_threshold=150M cp -r my_dir gs://my_bucket/my_dir
-## recursively copy a directory from your bucket to an existing directory on Sumner:
+//recursively copy a directory from your bucket to an existing directory on Sumner:
gsutil -m -o GSUtil:parallel_composite_upload_threshold=150M cp -r gs://my_bucket/my_dir my_dir
From 217e202cab3264c9d2d4cafe80b2476a2d837a85 Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi
Date: Mon, 22 Nov 2021 17:40:25 +0200
Subject: [PATCH 41/42] Fix: Removes rmats container from google.config
---
conf/executors/google.config | 1 -
1 file changed, 1 deletion(-)
diff --git a/conf/executors/google.config b/conf/executors/google.config
index 4200f406..6ce56876 100755
--- a/conf/executors/google.config
+++ b/conf/executors/google.config
@@ -82,7 +82,6 @@ process {
disk = params.gc_disk_size
cpus = { check_max (30 * task.attempt, 'cpus')}
memory = { check_max( 120.GB * task.attempt, 'memory' ) }
- container = 'gcr.io/nextflow-250616/rmats:4.1.0'
}
withName: 'paired_rmats' {
disk = params.gc_disk_size
From bfcf206047a0209f0fdcc2fdf14bc393f84040f9 Mon Sep 17 00:00:00 2001
From: Vlad-Dembrovskyi <64809705+Vlad-Dembrovskyi@users.noreply.github.com>
Date: Wed, 24 Nov 2021 20:51:16 +0200
Subject: [PATCH 42/42] Update changelog.md for v2.0
---
changelog.md | 39 +++++++++++++++++++++++----------------
1 file changed, 23 insertions(+), 16 deletions(-)
diff --git a/changelog.md b/changelog.md
index 0469f759..074cdcce 100644
--- a/changelog.md
+++ b/changelog.md
@@ -1,13 +1,24 @@
# Changelog
-## v 1.1 - Pipeline improvements
+### v2.0 - Pipeline improvements
-### Fixes:
- - Added missing trimmomatic logs to the multiqc report
- - Implemented correct support for input strandness in star process when `--stranded` is `second-strand` (was hardcoded to `strType=2` and only supported `first-strand` or `false` before)
+#### Improvements:
+ - Adds saving of all the process .command* log files to results/process-logs folder (#251)
+ - Adds pipeline workdir `--cleanup` option to clear all intermediate files on pipeline successful completion (true by default, false for CloudOS) (#238, #284, [089d6e3](https://github.com/TheJacksonLaboratory/splicing-pipelines-nf/pull/245/commits/3b71e038b186bb2bc92debacb02aede7b5dae917))
+ - Adds pipeline `--error_strategy` parameter to be able to specify pipeline error strategy directly from command line (doesn't work if specified in config linked by `-c` or `-config` nextflow params) (#267)
+ - Parametrizes google executor parameters so that pipeline can now be run on different CloudOS environments (#281)
+ - Adds a new `--download_from` option `FTP` mode to download SRA samples from [EBI FTP](https://ftp.sra.ebi.ac.uk/vol1/fastq/) (#283)
+- Adds new parameter `--save_unmapped` that makes saving of STAR unmapped files optional (false by default) (#284)
-### Updates:
- - Updated the following tools:
+#### Fixes:
+ - Adds missing trimmomatic logs to the multiqc report (#244)
+ - Implemented correct support for input strandness in star process when `--stranded` is `second-strand` (was hardcoded to `strType=2` and only supported `first-strand` or `false` before) (#264)
+ - Issue that stringti_merged results folder as well as some other folders are missing all or some files (#263)
+ - Fix pipeline crash when `params.stranded` was set to `false` (#276)
+ - Fixes old parameters in google.config that were undesirably overwriting nextflow.config parameters on CloudOS (#281, [217e202](https://github.com/TheJacksonLaboratory/splicing-pipelines-nf/pull/245/commits/217e202cab3264c9d2d4cafe80b2476a2d837a85))
+
+#### Updates:
+ - Updates the following tools: (#248)
- **STAR** `2.7.3` -> `2.7.9a` NOTE: Requires a new index! (updated in test profile)
- **Samtools** `1.10` -> `1.13`
- **StringTie** `2.1.3b` -> `2.1.7`
@@ -20,15 +31,11 @@
- sra-tools `2.10.8` -> `2.11.0`
- pigz `2.3.4` -> `2.6.0`
- gdc-client `1.5.0` -> `1.6.1`
- - Moved all containers to https://hub.docker.com/u/anczukowlab
+ - Moves all containers to https://hub.docker.com/u/anczukowlab
-### Maintenance:
- - Consideably reduced number of basic redundant CI tests by removing completely the `max_retries` matrix and `push` from `on: [push, pull_request]`
- - Added CI test for sra-downloading pipeline pathway (only supported with docker profile for now)
+#### Maintenance:
+ - Consideably reduces number of basic redundant CI tests by removing completely the `max_retries` matrix and `push` from `on: [push, pull_request]`
+ - Adds CI test for sra-downloading pipeline pathway (only supported with docker profile for now) (#253)
-### Enhancements:
- - Added saving of all the process .command* log files to results/process-logs folder
- - Added pipeline workdir `--cleanup` option to clear all intermediate files on pipeline successful completion
- - Added pipeline `--error_strategy` parameter to be able to specify pipeline error strategy directly from command line (doesn't work if specified in config linked by `-c` or `-config` nextflow params)
-
-## v 1.0 - Initial pipeline release
\ No newline at end of file
+
+## v 1.0 - Initial pipeline release