 +
+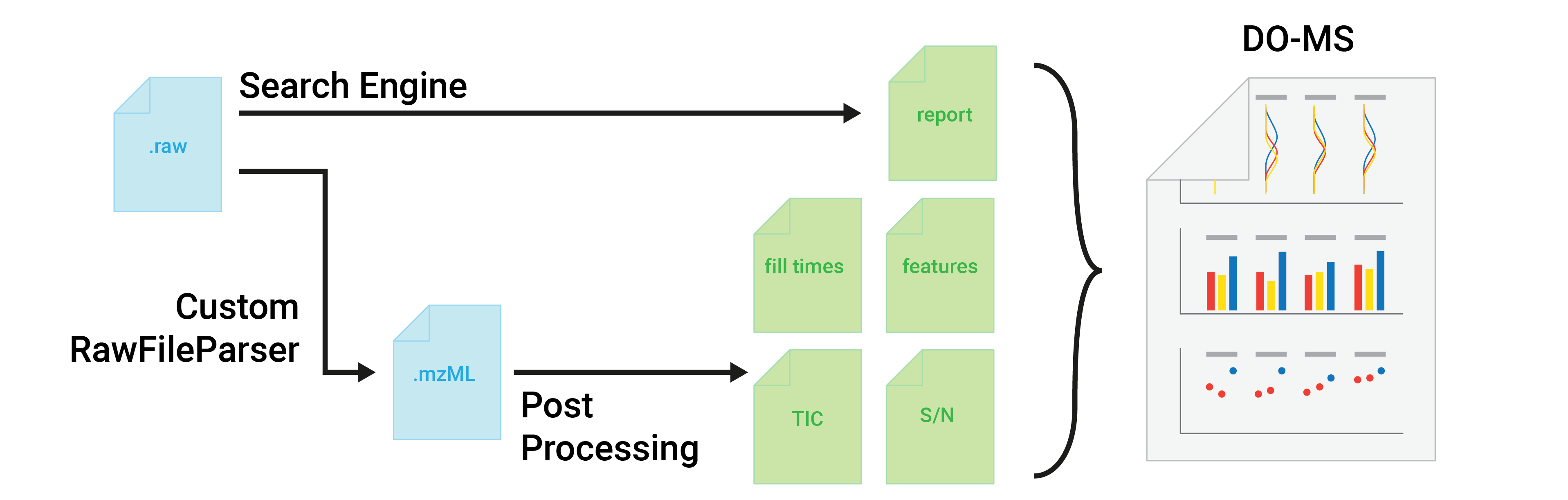 ## Getting Started
Please read our detailed getting started guides:
-* [Getting started on the application](https://do-ms.slavovlab.net/docs/getting-started-application)
-* [Getting started on the command-line](https://do-ms.slavovlab.net/docs/getting-started-command-line)
+* [Getting started with DIA preprocessing](https://do-ms.slavovlab.net/docs/getting-started-preprocessing)
+* [Getting started with DIA reports](https://do-ms.slavovlab.net/docs/getting-started-dia-app)
+* [Getting started with DDA reports](https://do-ms.slavovlab.net/docs/getting-started-dda-app)
### Requirements
-
-This application has been tested on R >= 4.0.0 and R <= 4.0.2, OSX 10.14 / Windows 7/8/10. R can be downloaded from the main [R Project page](https://www.r-project.org/) or downloaded with the [RStudio Application](https://www.rstudio.com/products/rstudio/download/). All modules are maintained for MaxQuant >= 1.6.0.16 and MaxQuant < 2.0
+This application has been tested on R >= 3.5.0, OSX 10.14 / Windows 7/8/10/11. R can be downloaded from the main [R Project page](https://www.r-project.org/) or downloaded with the [RStudio Application](https://www.rstudio.com/products/rstudio/download/). All modules are maintained for MaxQuant >= 1.6.0.16 and DIA-NN > 1.8.1.
The application suffers from visual glitches when displayed on unsupported older browsers (such as IE9 commonly packaged with RStudio on Windows). Please use IE >= 11, Firefox, or Chrome for the best user experience.
-### Installation
-
-Install this application by downloading it from the [release page](https://github.com/SlavovLab/DO-MS/releases/latest).
-
-### Running
+### Running the Interactive Application
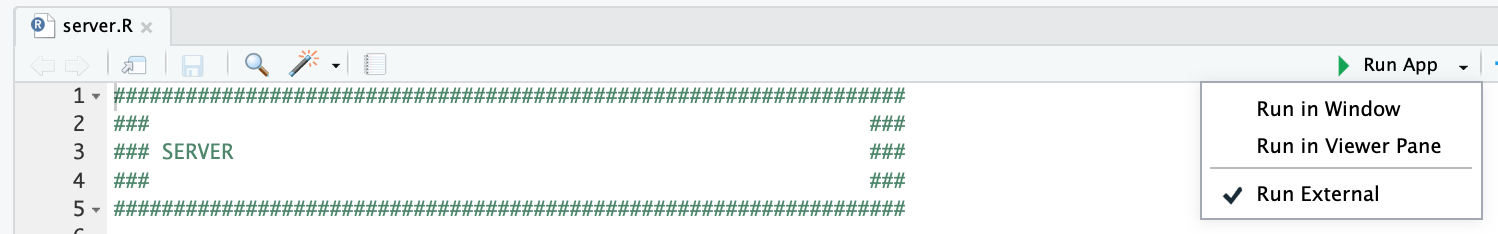
The easiest way to run the app is directly through RStudio, by opening the `DO-MS.Rproj` Rproject file
-
## Getting Started
Please read our detailed getting started guides:
-* [Getting started on the application](https://do-ms.slavovlab.net/docs/getting-started-application)
-* [Getting started on the command-line](https://do-ms.slavovlab.net/docs/getting-started-command-line)
+* [Getting started with DIA preprocessing](https://do-ms.slavovlab.net/docs/getting-started-preprocessing)
+* [Getting started with DIA reports](https://do-ms.slavovlab.net/docs/getting-started-dia-app)
+* [Getting started with DDA reports](https://do-ms.slavovlab.net/docs/getting-started-dda-app)
### Requirements
-
-This application has been tested on R >= 4.0.0 and R <= 4.0.2, OSX 10.14 / Windows 7/8/10. R can be downloaded from the main [R Project page](https://www.r-project.org/) or downloaded with the [RStudio Application](https://www.rstudio.com/products/rstudio/download/). All modules are maintained for MaxQuant >= 1.6.0.16 and MaxQuant < 2.0
+This application has been tested on R >= 3.5.0, OSX 10.14 / Windows 7/8/10/11. R can be downloaded from the main [R Project page](https://www.r-project.org/) or downloaded with the [RStudio Application](https://www.rstudio.com/products/rstudio/download/). All modules are maintained for MaxQuant >= 1.6.0.16 and DIA-NN > 1.8.1.
The application suffers from visual glitches when displayed on unsupported older browsers (such as IE9 commonly packaged with RStudio on Windows). Please use IE >= 11, Firefox, or Chrome for the best user experience.
-### Installation
-
-Install this application by downloading it from the [release page](https://github.com/SlavovLab/DO-MS/releases/latest).
-
-### Running
+### Running the Interactive Application
The easiest way to run the app is directly through RStudio, by opening the `DO-MS.Rproj` Rproject file
- +{: width="70%" .center-image}
and clicking the "Run App" button at the top of the application, after opening the `server.R` file. We recommend checking the "Run External" option to open the application in your default browser instead of the RStudio Viewer.
-
+{: width="70%" .center-image}
and clicking the "Run App" button at the top of the application, after opening the `server.R` file. We recommend checking the "Run External" option to open the application in your default browser instead of the RStudio Viewer.
- +{: width="70%" .center-image}
You can also start the application by running the `start_server.R` script.
-### Automated Report Generation
-
-You can automatically generate PDF/HTML reports without having to launch the server by running the `do-ms_cmd.R` script, like so:
-
-```
-$ Rscript do-ms_cmd.R config_file.yaml
-```
-
-This requires a configuration file, and you can [find an example one here](https://github.com/SlavovLab/DO-MS/blob/master/example/config_file.yaml). See [Automating Report Generation](https://do-ms.slavovlab.net/docs/automation) for more details and instructions.
-
### Customization
DO-MS is designed to be easily user-customizable for in-house proteomics workflows. Please see [Building Your Own Modules](https://do-ms.slavovlab.net/docs/build-your-own) for more details.
@@ -59,9 +46,9 @@ DO-MS is designed to be easily user-customizable for in-house proteomics workflo
Please see [Hosting as a Server](https://do-ms.slavovlab.net/docs/hosting-as-server) for more details.
-### Search Engines Other Than MaxQuant
+### Supporting other Search Engines
-This application is currently maintained for MaxQuant >= 1.6.0.16. Adapting to other search engines is possible but not provided out-of-the-box. Please see [Integrating Other Search Engines ](https://do-ms.slavovlab.net/docs/other-search-engines) for more details.
+This application is currently maintained for (MaxQuant)[https://www.nature.com/articles/nbt.1511] >= 1.6.0.16 and (DIA-NN)[https://www.nature.com/articles/s41592-019-0638-x] >= 1.8. Adapting to other search engines is possible but not provided out-of-the-box. Please see [Integrating Other Search Engines ](https://do-ms.slavovlab.net/docs/other-search-engines) for more details.
### Can I use this for Metabolomics, Lipidomics, etc... ?
@@ -71,13 +58,8 @@ While the base library of modules are based around bottom-up proteomics by LC-MS
## About the project
-
-
The manuscript for this tool is published at the Journal of Proteome Research: [https://pubs.acs.org/doi/10.1021/acs.jproteome.9b00039](https://pubs.acs.org/doi/10.1021/acs.jproteome.9b00039)
-
-The manuscript is also freely available on bioRxiv: [https://www.biorxiv.org/content/10.1101/512152v1](https://www.biorxiv.org/content/10.1101/512152v1).
+The manuscript for the extended version 2.0 can be found on bioArxiv: [https://www.biorxiv.org/content/10.1101/2023.02.02.526809v1](https://www.biorxiv.org/content/10.1101/2023.02.02.526809v1)
Contact the authors by email: [nslavov\{at\}northeastern.edu](mailto:nslavov@northeastern.edu).
@@ -87,21 +69,11 @@ DO-MS is distributed by an [MIT license](https://github.com/SlavovLab/DO-MS/blob
### Contributing
-Please feel free to contribute to this project by opening an issue or pull request.
-
-
-
-
+Please feel free to contribute to this project by opening an issue or pull request in the [GitHub repository](https://github.com/SlavovLab/DO-MS).
-------------
## Help!
-For any bugs, questions, or feature requests,
+For any bugs, questions, or feature requests,
please use the [GitHub issue system](https://github.com/SlavovLab/DO-MS/issues) to contact the developers.
diff --git a/do-ms_cmd.R b/do-ms_cmd.R
old mode 100755
new mode 100644
index 9a53d1c..033e884
--- a/do-ms_cmd.R
+++ b/do-ms_cmd.R
@@ -244,7 +244,7 @@ for(i in 1:length(config[['load_misc_input_files']])) {
prnt(paste0('Loading misc file: ', name))
# read in as data frame (need to convert from tibble)
- data[[name]] <- as.data.frame(read_tsv(file=path))
+ data[[name]] <- as.data.frame(read_tsv(file=path, col_types = cols()))
# rename columns (replace whitespace or special characters with '.')
colnames(data[[name]]) <- gsub('\\s|\\(|\\)|\\/|\\[|\\]', '.',
colnames(data[[name]]))
@@ -363,6 +363,8 @@ for(f in config[['load_input_files']]) {
# sort the raw files
raw_files <- sort(raw_files)
+
+
# load naming format
file_levels <- rep(config[['exp_name_format']], length(raw_files))
@@ -377,8 +379,12 @@ file_levels <- str_replace(file_levels, '\\%i', as.character(seq(1, length(raw_f
# folder name is stored as the names of the raw files vector
file_levels <- str_replace(file_levels, '\\%f', names(raw_files))
+
# replace %e with the raw file name
file_levels <- str_replace(file_levels, '\\%e', raw_files)
+print(file_levels)
+
+print(config[['exp_name_pattern']])
# apply custom string extraction expression to file levels
if(!is.null(config[['exp_name_pattern']])) {
@@ -387,11 +393,13 @@ if(!is.null(config[['exp_name_pattern']])) {
file_levels[is.na(file_levels)] <- 'default'
}
+print(file_levels)
+
# apply custom names, as defined in the "exp_names" config field
if(!is.null(config[['exp_names']]) & length(config[['exp_names']]) > 0) {
file_levels[1:length(config[['exp_names']])] <- config[['exp_names']]
}
-
+print(file_levels)
# ensure there are no duplicate names
# if so, then append a suffix to duplicate names to prevent refactoring errors
@@ -477,4 +485,4 @@ generate_report(input, f_data, raw_files, config[['output']], progress_bar=FALSE
# prnt(paste0('Report written to: ', config[['output']]))
-prnt('Done!')
+prnt('Done!')
\ No newline at end of file
diff --git a/docs/Gemfile b/docs/Gemfile
index 3be9c3c..457656e 100644
--- a/docs/Gemfile
+++ b/docs/Gemfile
@@ -1,2 +1,6 @@
source "https://rubygems.org"
gemspec
+
+gem "rexml", "~> 3.2"
+
+gem "webrick", "~> 1.8"
diff --git a/docs/_config.yml b/docs/_config.yml
index b3ce700..3eccc36 100644
--- a/docs/_config.yml
+++ b/docs/_config.yml
@@ -26,17 +26,15 @@ search_enabled: true
# Aux links for the upper right navigation
aux_links:
- "DO-MS Preprint":
- - "https://www.biorxiv.org/content/10.1101/512152v1"
- "JPR Article":
- - "https://pubs.acs.org/doi/10.1021/acs.jproteome.9b00039"
+ "BioArxiv Preprint":
+ - "https://www.biorxiv.org/content/10.1101/2023.02.02.526809v1"
"GitHub Repository":
- "https://github.com/SlavovLab/DO-MS"
"Slavov Lab":
- "http://slavovlab.net"
github_link: https://github.com/SlavovLab/DO-MS
-preprint_link: https://www.biorxiv.org/content/10.1101/512152v1
+preprint_link: https://www.biorxiv.org/content/10.1101/2023.02.02.526809v1
jpr_link: https://pubs.acs.org/doi/10.1021/acs.jproteome.9b00039
# Color scheme currently only supports "dark" or nil (default)
diff --git a/docs/assets/images/dia_input.png b/docs/assets/images/dia_input.png
new file mode 100644
index 0000000..b9b341c
Binary files /dev/null and b/docs/assets/images/dia_input.png differ
diff --git a/docs/assets/images/do-ms-dia-example.png b/docs/assets/images/do-ms-dia-example.png
new file mode 100644
index 0000000..8d4ae3f
Binary files /dev/null and b/docs/assets/images/do-ms-dia-example.png differ
diff --git a/docs/assets/images/do-ms-dia-foldername.png b/docs/assets/images/do-ms-dia-foldername.png
new file mode 100644
index 0000000..cf4b699
Binary files /dev/null and b/docs/assets/images/do-ms-dia-foldername.png differ
diff --git a/docs/assets/images/do-ms-dia-generate-report.png b/docs/assets/images/do-ms-dia-generate-report.png
new file mode 100644
index 0000000..b9632e3
Binary files /dev/null and b/docs/assets/images/do-ms-dia-generate-report.png differ
diff --git a/docs/assets/images/do-ms-dia-import.png b/docs/assets/images/do-ms-dia-import.png
new file mode 100644
index 0000000..e4d42a6
Binary files /dev/null and b/docs/assets/images/do-ms-dia-import.png differ
diff --git a/docs/assets/images/do-ms-dia-info.png b/docs/assets/images/do-ms-dia-info.png
new file mode 100644
index 0000000..a77dbbb
Binary files /dev/null and b/docs/assets/images/do-ms-dia-info.png differ
diff --git a/docs/assets/images/do-ms-dia-load.png b/docs/assets/images/do-ms-dia-load.png
new file mode 100644
index 0000000..5c2decb
Binary files /dev/null and b/docs/assets/images/do-ms-dia-load.png differ
diff --git a/docs/assets/images/do-ms-dia-overview.png b/docs/assets/images/do-ms-dia-overview.png
new file mode 100644
index 0000000..6079f1c
Binary files /dev/null and b/docs/assets/images/do-ms-dia-overview.png differ
diff --git a/docs/assets/images/do-ms-dia-pathname.png b/docs/assets/images/do-ms-dia-pathname.png
new file mode 100644
index 0000000..b4b2b57
Binary files /dev/null and b/docs/assets/images/do-ms-dia-pathname.png differ
diff --git a/docs/assets/images/do-ms-dia-rename.png b/docs/assets/images/do-ms-dia-rename.png
new file mode 100644
index 0000000..debb1eb
Binary files /dev/null and b/docs/assets/images/do-ms-dia-rename.png differ
diff --git a/docs/assets/images/do-ms-dia-results.png b/docs/assets/images/do-ms-dia-results.png
new file mode 100644
index 0000000..a130b41
Binary files /dev/null and b/docs/assets/images/do-ms-dia-results.png differ
diff --git a/docs/assets/images/do-ms-dia_config.png b/docs/assets/images/do-ms-dia_config.png
new file mode 100644
index 0000000..1d79228
Binary files /dev/null and b/docs/assets/images/do-ms-dia_config.png differ
diff --git a/docs/assets/images/do-ms-dia_mode.png b/docs/assets/images/do-ms-dia_mode.png
new file mode 100644
index 0000000..0ab4770
Binary files /dev/null and b/docs/assets/images/do-ms-dia_mode.png differ
diff --git a/docs/assets/images/do-ms-dia_title.png b/docs/assets/images/do-ms-dia_title.png
new file mode 100755
index 0000000..276d1d2
Binary files /dev/null and b/docs/assets/images/do-ms-dia_title.png differ
diff --git a/docs/assets/images/do-ms-dia_title_v2.png b/docs/assets/images/do-ms-dia_title_v2.png
new file mode 100644
index 0000000..9226b55
Binary files /dev/null and b/docs/assets/images/do-ms-dia_title_v2.png differ
diff --git a/docs/assets/images/preprocessing_input.png b/docs/assets/images/preprocessing_input.png
new file mode 100644
index 0000000..49ac6e3
Binary files /dev/null and b/docs/assets/images/preprocessing_input.png differ
diff --git a/docs/assets/images/preprocessing_output.png b/docs/assets/images/preprocessing_output.png
new file mode 100644
index 0000000..a70bd6b
Binary files /dev/null and b/docs/assets/images/preprocessing_output.png differ
diff --git a/docs/docs/DO-MS_examples.md b/docs/docs/DO-MS_examples.md
index 2e85552..1de564b 100644
--- a/docs/docs/DO-MS_examples.md
+++ b/docs/docs/DO-MS_examples.md
@@ -1,7 +1,7 @@
---
layout: default
title: DO-MS Examples
-nav_order: 3
+nav_order: 5
permalink: docs/DO-MS_examples
parent: Getting Started
---
@@ -10,6 +10,13 @@ parent: Getting Started
Bellow are links to example DO-MS Reports
+## DIA-NN DIA Reports
+1. [MS2 number optimization](https://drive.google.com/uc?id=1mNrJsk6uaI3ljtQEadr0XwR5mlyO4xZg&export=download)
+2. [Different MS2 strategies](https://drive.google.com/uc?id=1eOJXC2Zb0lmVwbsaj7Zlf6wm3SPGrbWl&export=download)
+3. [Gradient length optimization](https://drive.google.com/uc?id=1dQdo3kR-WxWT8zEeOxztxXmJE1rSMsEM&export=download)
+4. [MS1 sampling optimization](https://drive.google.com/uc?id=1YcT3al9ICu36_ornSsLZgHj1eTBBPOxb&export=download)
+5. [Single cell quality control](https://drive.google.com/uc?id=1v62qJ8JVZ_TSkUa3K0i6nWoJDbW9EQHO&export=download)
+## MaxQuant DDA Reports
1. [Isobaric carrier optimization](https://scope2.slavovlab.net/mass-spec/Isobaric-carrier-optimization#do-ms-reports)
2. [Technical improvements of SCoPE2](https://scope2.slavovlab.net/mass-spec/Increased-accuracy-and-throughput)
diff --git a/docs/docs/getting_started.md b/docs/docs/getting_started.md
index ea238bb..290e93d 100644
--- a/docs/docs/getting_started.md
+++ b/docs/docs/getting_started.md
@@ -8,4 +8,19 @@ has_children: true
# Getting Started
-DO-MS can be run either from the command-line or as interactive application. Follow the links below to get started using the implementation of your choice. For more details on the data display, read the [DO-MS article](https://pubs.acs.org/doi/10.1021/acs.jproteome.9b00039).
+DO-MS can be run either from the command-line or as interactive application. Follow the links below to get started using the implementation of your choice. For more details on the data display, read the [DO-MS 2.0 article](https://www.biorxiv.org/content/10.1101/2023.02.02.526809v1).
+
+Before starting DO-MS the first time, the input data type has to be selected. DO-MS can work with both DDA results coming from MaxQuant as well as with DIA results coming from DIA-NN.
+The mode can be set in the config.yaml file. Open the file in R-Studio or your editor of choice.
+{: width="70%" .center-image}
+
+If You wish to analyze MaxQuant DDA results, change the parameter to `max_quant`, otherwise leave it as `dia-nn`. This setting needs to be changed before SO-MS is started or the R environment is initialized. It is also possible to keep both versionas in two separate folders simultanously.
+{: width="70%" .center-image}
+
+# Generating DO-MS Reports
+Please read our detailed getting started guides:
+
+* [DIA Preprocessing]({{site.baseurl}}/docs/getting-started-preprocessing)
+* [DIA Reports using the app]({{site.baseurl}}/docs/getting-started-dia-app)
+* [DDA Reports using the app]({{site.baseurl}}/docs/getting-started-application)
+* [DDA Reports using the command line]({{site.baseurl}}/docs/getting-started-application)
\ No newline at end of file
diff --git a/docs/docs/getting_started_app.md b/docs/docs/getting_started_app.md
index e207619..3d6c702 100644
--- a/docs/docs/getting_started_app.md
+++ b/docs/docs/getting_started_app.md
@@ -1,12 +1,12 @@
---
layout: default
-title: Interactive Application
-nav_order: 1
-permalink: docs/getting-started-application
+title: DDA Reports in the App
+nav_order: 3
+permalink: docs/getting-started-dda-app
parent: Getting Started
---
-# Getting Started -- Interactive DO-MS Application
+# Getting Started -- DDA Reports in the App
DO-MS is an application to visualize mass-spec data both in an interactive application and static reports generated via. the command-line. In this document we'll walk you through analyzing an example dataset in the interactive application.
@@ -31,13 +31,8 @@ The only constraint for data in DO-MS is that it must be from MaxQuant version >
## Installation
-Download the application via. a zip or tar archive from the [GitHub release page](https://github.com/SlavovLab/DO-MS/releases). Unzip the archive, and then open the `DO-MS.Rproj` to load the project into RStudio
-
-{: width="60%" .center-image}
-
-To start the app open `server.R` in RStudio and on the top right corner of the editor click on the "Run App" button. To run the application in your browser (preferred option) rather than in RStudio, click the dropdown menu and select "Run External".
-
-{: width="85%" .center-image}
+Please make sure that you installed DO-MS as descibed in the [installation]({{site.baseurl}}/docs/installation) section.
+
+{: width="70%" .center-image}
You can also start the application by running the `start_server.R` script.
-### Automated Report Generation
-
-You can automatically generate PDF/HTML reports without having to launch the server by running the `do-ms_cmd.R` script, like so:
-
-```
-$ Rscript do-ms_cmd.R config_file.yaml
-```
-
-This requires a configuration file, and you can [find an example one here](https://github.com/SlavovLab/DO-MS/blob/master/example/config_file.yaml). See [Automating Report Generation](https://do-ms.slavovlab.net/docs/automation) for more details and instructions.
-
### Customization
DO-MS is designed to be easily user-customizable for in-house proteomics workflows. Please see [Building Your Own Modules](https://do-ms.slavovlab.net/docs/build-your-own) for more details.
@@ -59,9 +46,9 @@ DO-MS is designed to be easily user-customizable for in-house proteomics workflo
Please see [Hosting as a Server](https://do-ms.slavovlab.net/docs/hosting-as-server) for more details.
-### Search Engines Other Than MaxQuant
+### Supporting other Search Engines
-This application is currently maintained for MaxQuant >= 1.6.0.16. Adapting to other search engines is possible but not provided out-of-the-box. Please see [Integrating Other Search Engines ](https://do-ms.slavovlab.net/docs/other-search-engines) for more details.
+This application is currently maintained for (MaxQuant)[https://www.nature.com/articles/nbt.1511] >= 1.6.0.16 and (DIA-NN)[https://www.nature.com/articles/s41592-019-0638-x] >= 1.8. Adapting to other search engines is possible but not provided out-of-the-box. Please see [Integrating Other Search Engines ](https://do-ms.slavovlab.net/docs/other-search-engines) for more details.
### Can I use this for Metabolomics, Lipidomics, etc... ?
@@ -71,13 +58,8 @@ While the base library of modules are based around bottom-up proteomics by LC-MS
## About the project
-
-
The manuscript for this tool is published at the Journal of Proteome Research: [https://pubs.acs.org/doi/10.1021/acs.jproteome.9b00039](https://pubs.acs.org/doi/10.1021/acs.jproteome.9b00039)
-
-The manuscript is also freely available on bioRxiv: [https://www.biorxiv.org/content/10.1101/512152v1](https://www.biorxiv.org/content/10.1101/512152v1).
+The manuscript for the extended version 2.0 can be found on bioArxiv: [https://www.biorxiv.org/content/10.1101/2023.02.02.526809v1](https://www.biorxiv.org/content/10.1101/2023.02.02.526809v1)
Contact the authors by email: [nslavov\{at\}northeastern.edu](mailto:nslavov@northeastern.edu).
@@ -87,21 +69,11 @@ DO-MS is distributed by an [MIT license](https://github.com/SlavovLab/DO-MS/blob
### Contributing
-Please feel free to contribute to this project by opening an issue or pull request.
-
-
-
-
+Please feel free to contribute to this project by opening an issue or pull request in the [GitHub repository](https://github.com/SlavovLab/DO-MS).
-------------
## Help!
-For any bugs, questions, or feature requests,
+For any bugs, questions, or feature requests,
please use the [GitHub issue system](https://github.com/SlavovLab/DO-MS/issues) to contact the developers.
diff --git a/do-ms_cmd.R b/do-ms_cmd.R
old mode 100755
new mode 100644
index 9a53d1c..033e884
--- a/do-ms_cmd.R
+++ b/do-ms_cmd.R
@@ -244,7 +244,7 @@ for(i in 1:length(config[['load_misc_input_files']])) {
prnt(paste0('Loading misc file: ', name))
# read in as data frame (need to convert from tibble)
- data[[name]] <- as.data.frame(read_tsv(file=path))
+ data[[name]] <- as.data.frame(read_tsv(file=path, col_types = cols()))
# rename columns (replace whitespace or special characters with '.')
colnames(data[[name]]) <- gsub('\\s|\\(|\\)|\\/|\\[|\\]', '.',
colnames(data[[name]]))
@@ -363,6 +363,8 @@ for(f in config[['load_input_files']]) {
# sort the raw files
raw_files <- sort(raw_files)
+
+
# load naming format
file_levels <- rep(config[['exp_name_format']], length(raw_files))
@@ -377,8 +379,12 @@ file_levels <- str_replace(file_levels, '\\%i', as.character(seq(1, length(raw_f
# folder name is stored as the names of the raw files vector
file_levels <- str_replace(file_levels, '\\%f', names(raw_files))
+
# replace %e with the raw file name
file_levels <- str_replace(file_levels, '\\%e', raw_files)
+print(file_levels)
+
+print(config[['exp_name_pattern']])
# apply custom string extraction expression to file levels
if(!is.null(config[['exp_name_pattern']])) {
@@ -387,11 +393,13 @@ if(!is.null(config[['exp_name_pattern']])) {
file_levels[is.na(file_levels)] <- 'default'
}
+print(file_levels)
+
# apply custom names, as defined in the "exp_names" config field
if(!is.null(config[['exp_names']]) & length(config[['exp_names']]) > 0) {
file_levels[1:length(config[['exp_names']])] <- config[['exp_names']]
}
-
+print(file_levels)
# ensure there are no duplicate names
# if so, then append a suffix to duplicate names to prevent refactoring errors
@@ -477,4 +485,4 @@ generate_report(input, f_data, raw_files, config[['output']], progress_bar=FALSE
# prnt(paste0('Report written to: ', config[['output']]))
-prnt('Done!')
+prnt('Done!')
\ No newline at end of file
diff --git a/docs/Gemfile b/docs/Gemfile
index 3be9c3c..457656e 100644
--- a/docs/Gemfile
+++ b/docs/Gemfile
@@ -1,2 +1,6 @@
source "https://rubygems.org"
gemspec
+
+gem "rexml", "~> 3.2"
+
+gem "webrick", "~> 1.8"
diff --git a/docs/_config.yml b/docs/_config.yml
index b3ce700..3eccc36 100644
--- a/docs/_config.yml
+++ b/docs/_config.yml
@@ -26,17 +26,15 @@ search_enabled: true
# Aux links for the upper right navigation
aux_links:
- "DO-MS Preprint":
- - "https://www.biorxiv.org/content/10.1101/512152v1"
- "JPR Article":
- - "https://pubs.acs.org/doi/10.1021/acs.jproteome.9b00039"
+ "BioArxiv Preprint":
+ - "https://www.biorxiv.org/content/10.1101/2023.02.02.526809v1"
"GitHub Repository":
- "https://github.com/SlavovLab/DO-MS"
"Slavov Lab":
- "http://slavovlab.net"
github_link: https://github.com/SlavovLab/DO-MS
-preprint_link: https://www.biorxiv.org/content/10.1101/512152v1
+preprint_link: https://www.biorxiv.org/content/10.1101/2023.02.02.526809v1
jpr_link: https://pubs.acs.org/doi/10.1021/acs.jproteome.9b00039
# Color scheme currently only supports "dark" or nil (default)
diff --git a/docs/assets/images/dia_input.png b/docs/assets/images/dia_input.png
new file mode 100644
index 0000000..b9b341c
Binary files /dev/null and b/docs/assets/images/dia_input.png differ
diff --git a/docs/assets/images/do-ms-dia-example.png b/docs/assets/images/do-ms-dia-example.png
new file mode 100644
index 0000000..8d4ae3f
Binary files /dev/null and b/docs/assets/images/do-ms-dia-example.png differ
diff --git a/docs/assets/images/do-ms-dia-foldername.png b/docs/assets/images/do-ms-dia-foldername.png
new file mode 100644
index 0000000..cf4b699
Binary files /dev/null and b/docs/assets/images/do-ms-dia-foldername.png differ
diff --git a/docs/assets/images/do-ms-dia-generate-report.png b/docs/assets/images/do-ms-dia-generate-report.png
new file mode 100644
index 0000000..b9632e3
Binary files /dev/null and b/docs/assets/images/do-ms-dia-generate-report.png differ
diff --git a/docs/assets/images/do-ms-dia-import.png b/docs/assets/images/do-ms-dia-import.png
new file mode 100644
index 0000000..e4d42a6
Binary files /dev/null and b/docs/assets/images/do-ms-dia-import.png differ
diff --git a/docs/assets/images/do-ms-dia-info.png b/docs/assets/images/do-ms-dia-info.png
new file mode 100644
index 0000000..a77dbbb
Binary files /dev/null and b/docs/assets/images/do-ms-dia-info.png differ
diff --git a/docs/assets/images/do-ms-dia-load.png b/docs/assets/images/do-ms-dia-load.png
new file mode 100644
index 0000000..5c2decb
Binary files /dev/null and b/docs/assets/images/do-ms-dia-load.png differ
diff --git a/docs/assets/images/do-ms-dia-overview.png b/docs/assets/images/do-ms-dia-overview.png
new file mode 100644
index 0000000..6079f1c
Binary files /dev/null and b/docs/assets/images/do-ms-dia-overview.png differ
diff --git a/docs/assets/images/do-ms-dia-pathname.png b/docs/assets/images/do-ms-dia-pathname.png
new file mode 100644
index 0000000..b4b2b57
Binary files /dev/null and b/docs/assets/images/do-ms-dia-pathname.png differ
diff --git a/docs/assets/images/do-ms-dia-rename.png b/docs/assets/images/do-ms-dia-rename.png
new file mode 100644
index 0000000..debb1eb
Binary files /dev/null and b/docs/assets/images/do-ms-dia-rename.png differ
diff --git a/docs/assets/images/do-ms-dia-results.png b/docs/assets/images/do-ms-dia-results.png
new file mode 100644
index 0000000..a130b41
Binary files /dev/null and b/docs/assets/images/do-ms-dia-results.png differ
diff --git a/docs/assets/images/do-ms-dia_config.png b/docs/assets/images/do-ms-dia_config.png
new file mode 100644
index 0000000..1d79228
Binary files /dev/null and b/docs/assets/images/do-ms-dia_config.png differ
diff --git a/docs/assets/images/do-ms-dia_mode.png b/docs/assets/images/do-ms-dia_mode.png
new file mode 100644
index 0000000..0ab4770
Binary files /dev/null and b/docs/assets/images/do-ms-dia_mode.png differ
diff --git a/docs/assets/images/do-ms-dia_title.png b/docs/assets/images/do-ms-dia_title.png
new file mode 100755
index 0000000..276d1d2
Binary files /dev/null and b/docs/assets/images/do-ms-dia_title.png differ
diff --git a/docs/assets/images/do-ms-dia_title_v2.png b/docs/assets/images/do-ms-dia_title_v2.png
new file mode 100644
index 0000000..9226b55
Binary files /dev/null and b/docs/assets/images/do-ms-dia_title_v2.png differ
diff --git a/docs/assets/images/preprocessing_input.png b/docs/assets/images/preprocessing_input.png
new file mode 100644
index 0000000..49ac6e3
Binary files /dev/null and b/docs/assets/images/preprocessing_input.png differ
diff --git a/docs/assets/images/preprocessing_output.png b/docs/assets/images/preprocessing_output.png
new file mode 100644
index 0000000..a70bd6b
Binary files /dev/null and b/docs/assets/images/preprocessing_output.png differ
diff --git a/docs/docs/DO-MS_examples.md b/docs/docs/DO-MS_examples.md
index 2e85552..1de564b 100644
--- a/docs/docs/DO-MS_examples.md
+++ b/docs/docs/DO-MS_examples.md
@@ -1,7 +1,7 @@
---
layout: default
title: DO-MS Examples
-nav_order: 3
+nav_order: 5
permalink: docs/DO-MS_examples
parent: Getting Started
---
@@ -10,6 +10,13 @@ parent: Getting Started
Bellow are links to example DO-MS Reports
+## DIA-NN DIA Reports
+1. [MS2 number optimization](https://drive.google.com/uc?id=1mNrJsk6uaI3ljtQEadr0XwR5mlyO4xZg&export=download)
+2. [Different MS2 strategies](https://drive.google.com/uc?id=1eOJXC2Zb0lmVwbsaj7Zlf6wm3SPGrbWl&export=download)
+3. [Gradient length optimization](https://drive.google.com/uc?id=1dQdo3kR-WxWT8zEeOxztxXmJE1rSMsEM&export=download)
+4. [MS1 sampling optimization](https://drive.google.com/uc?id=1YcT3al9ICu36_ornSsLZgHj1eTBBPOxb&export=download)
+5. [Single cell quality control](https://drive.google.com/uc?id=1v62qJ8JVZ_TSkUa3K0i6nWoJDbW9EQHO&export=download)
+## MaxQuant DDA Reports
1. [Isobaric carrier optimization](https://scope2.slavovlab.net/mass-spec/Isobaric-carrier-optimization#do-ms-reports)
2. [Technical improvements of SCoPE2](https://scope2.slavovlab.net/mass-spec/Increased-accuracy-and-throughput)
diff --git a/docs/docs/getting_started.md b/docs/docs/getting_started.md
index ea238bb..290e93d 100644
--- a/docs/docs/getting_started.md
+++ b/docs/docs/getting_started.md
@@ -8,4 +8,19 @@ has_children: true
# Getting Started
-DO-MS can be run either from the command-line or as interactive application. Follow the links below to get started using the implementation of your choice. For more details on the data display, read the [DO-MS article](https://pubs.acs.org/doi/10.1021/acs.jproteome.9b00039).
+DO-MS can be run either from the command-line or as interactive application. Follow the links below to get started using the implementation of your choice. For more details on the data display, read the [DO-MS 2.0 article](https://www.biorxiv.org/content/10.1101/2023.02.02.526809v1).
+
+Before starting DO-MS the first time, the input data type has to be selected. DO-MS can work with both DDA results coming from MaxQuant as well as with DIA results coming from DIA-NN.
+The mode can be set in the config.yaml file. Open the file in R-Studio or your editor of choice.
+{: width="70%" .center-image}
+
+If You wish to analyze MaxQuant DDA results, change the parameter to `max_quant`, otherwise leave it as `dia-nn`. This setting needs to be changed before SO-MS is started or the R environment is initialized. It is also possible to keep both versionas in two separate folders simultanously.
+{: width="70%" .center-image}
+
+# Generating DO-MS Reports
+Please read our detailed getting started guides:
+
+* [DIA Preprocessing]({{site.baseurl}}/docs/getting-started-preprocessing)
+* [DIA Reports using the app]({{site.baseurl}}/docs/getting-started-dia-app)
+* [DDA Reports using the app]({{site.baseurl}}/docs/getting-started-application)
+* [DDA Reports using the command line]({{site.baseurl}}/docs/getting-started-application)
\ No newline at end of file
diff --git a/docs/docs/getting_started_app.md b/docs/docs/getting_started_app.md
index e207619..3d6c702 100644
--- a/docs/docs/getting_started_app.md
+++ b/docs/docs/getting_started_app.md
@@ -1,12 +1,12 @@
---
layout: default
-title: Interactive Application
-nav_order: 1
-permalink: docs/getting-started-application
+title: DDA Reports in the App
+nav_order: 3
+permalink: docs/getting-started-dda-app
parent: Getting Started
---
-# Getting Started -- Interactive DO-MS Application
+# Getting Started -- DDA Reports in the App
DO-MS is an application to visualize mass-spec data both in an interactive application and static reports generated via. the command-line. In this document we'll walk you through analyzing an example dataset in the interactive application.
@@ -31,13 +31,8 @@ The only constraint for data in DO-MS is that it must be from MaxQuant version >
## Installation
-Download the application via. a zip or tar archive from the [GitHub release page](https://github.com/SlavovLab/DO-MS/releases). Unzip the archive, and then open the `DO-MS.Rproj` to load the project into RStudio
-
-{: width="60%" .center-image}
-
-To start the app open `server.R` in RStudio and on the top right corner of the editor click on the "Run App" button. To run the application in your browser (preferred option) rather than in RStudio, click the dropdown menu and select "Run External".
-
-{: width="85%" .center-image}
+Please make sure that you installed DO-MS as descibed in the [installation]({{site.baseurl}}/docs/installation) section.
+DO-MS Report
+Version: 2.0.b5 | PEP < -Inf | Generated: 2022-05-18 18:22:32
+ +Summary
+DIA-NN Experiments
+DIA-NN Experiments
+wGW032 F:/GW/raw_data/wGW032.raw
+wGW033 F:/GW/raw_data/wGW033.raw
+wGW034 F:/GW/raw_data/wGW034.raw
+wGW035 F:/GW/raw_data/wGW035.raw
+wGW036 F:/GW/raw_data/wGW036.raw
+wGW037 F:/GW/raw_data/wGW037.raw
+wGW038 F:/GW/raw_data/wGW038.raw
+Ion Sampling
+Channel wise MS1 Intensity for Precursors
+Plotting the MS1 intensity for all precursors which were associated with one of the defined channels.
+## [1] "Plot failed to render. Reason: Error in `[.data.frame`(data()[[\"report\"]], , c(\"Raw.file\", \"Ms1.Area\", : undefined columns selected\n"Precursors Identified across Gradient
+Precursor are plotted across the chromatographic gradient.
+
MS1 Intensity summed over all Channels.
+Plotting the MS1 intensity for all precursors summed over all channels.
+## [1] "Plot failed to render. Reason: Error in `[.data.frame`(data()[[\"report\"]], , c(\"Raw.file\", \"Ms1.Area\", : undefined columns selected\n"MS1 Intensity for Intersected Precursors, summed over all Channels.
+Plotting the MS1 intensity for all precursors summed over all channels. Only intersected precursors present in all loaded experiments are shown.
+
Normalized MS1 Intensity for Intersected Precursors
+Plotting the MS1 Intensity for intersected precursors summed over all channels. Experiments are normalized to the first experiment.
+
Number of Precursors by Charge State
+Number of precursors observed during MS1 scans by charge state
+
Ms1 Fill Time Distribution
+Ms1 fill times along gradient
+## [1] "Plot failed to render. Reason: Error: Upload fill_times.txt\n"Ms1 Fill Times along Gradient
+The averge fill time is shown in magenta for different bins along the retention time gradient. The standard deviation is depicted as area in blue, scans outside this area are shown as single datapoints.
+## [1] "Plot failed to render. Reason: Error: Upload fill_times.txt\n"Ms1 total Ion Current along Gradient
+The toal Ion Current (TIC) is shown for bins along the retention time gradient.
+## [1] "Plot failed to render. Reason: Error: Upload tic.tsv\n"Ms2 Fill Time Distribution
+Ms2 fill times along gradient
+## [1] "Plot failed to render. Reason: Error: Upload fill_times.txt\n"Ms2 Fill Times along Gradient
+The averge fill time is shown in magenta for different bins along the retention time gradient. The standard deviation is depicted as area in blue, scans outside this area are shown as single datapoints.
+## [1] "Plot failed to render. Reason: Error: Upload fill_times.txt\n"Ms2 Fill Time Matrix
+The average Ms2 fill times are shown across the gradient for every distinct Ms2 window.
+## [1] "Plot failed to render. Reason: Error: Upload tic.tsv\n"Channel wise MS1 Counts for Precursors
+Plotting the MS1 counts based on the signal to noise ratio for all precursors which were associated with one of the defined channels.
+## [1] "Plot failed to render. Reason: Error: Upload report.txt\n"Identifications
+Number of Confident Precursor Identifications
+Plotting the number of precursors identified at each given confidence level.
+
Precursors by Quantification Strategy
+Number of precursors found based on quantification startegy. Ms2 precursors are counted based on Precursor.Quantity > 0 and Ms1 precursors are counted based on Ms1.Area > 0.
+
Precursors by Modification
+Number of precursors found based on modification types specified
+## [1] "Plot failed to render. Reason: Error in `[.data.frame`(data()[[\"report\"]], , c(\"Raw.file\", \"Ms1.Area\", : undefined columns selected\n"Miscleavage Rate (percentage), PEP < 0.01
+Miscleavage rate (percentage) for precursors identified with confidence PEP < 0.01
+Miscleavage Rate (K), PEP < 0.01
+Plotting frequency of lysine miscleavages in confidently identified precursors.
+
Miscleavage Rate (R), PEP < 0.01
+Plotting frequency of arginine miscleavages in confidently identified precursors.
+
Number of Protein Identifications
+Number of proteotypic protein IDs found per run. Protein IDs are shown across all channels in an experiment.
+## [1] "Plot failed to render. Reason: Error in `[.data.frame`(data()[[\"report\"]], , c(\"Raw.file\", \"Ms1.Area\", : undefined columns selected\n"plex-DIA Diagnostics
+Identified Precursors per Channel
+The number of precursors identified is shown for every channel together with the number of total and intersected precursors. The number of precursors is based on all precursors found in the report.tsv file which is by default controlled by a run-specific FDR.
+## [1] "Plot failed to render. Reason: Error in `[.data.frame`(data()[[\"report\"]], , c(\"Raw.file\", \"Ms1.Area\", : undefined columns selected\n"Identified Precursors per Channel, Channel Q-Value
+The number of precursors identified is shown for every channel together with the number of total and intersected precursors. The number of precursors is based on all precursors with a Channel.Q.Value <= 0.01.
+## [1] "Plot failed to render. Reason: Error in `[.data.frame`(data()[[\"report\"]], , c(\"Raw.file\", \"Ms1.Area\", : undefined columns selected\n"Identified Proteins per Channel
+The number of Proteins identified is shown for every channel together with the number of total and intersected proteins. The number of proteins is based on all proteotypic precursors independent of the Protein.Q.Value.
+## [1] "Plot failed to render. Reason: Error in `[.data.frame`(data()[[\"report\"]], , c(\"Raw.file\", \"Ms1.Area\", : undefined columns selected\n"Relative Single-Cell Intensity
+Single-Cell intensity relative to the carrier channel for intersected precursors
+## [1] "Plot failed to render. Reason: Error in `[.data.frame`(data()[[\"report\"]], , c(\"Raw.file\", \"Ms1.Area\", : undefined columns selected\n"Total MS1 Precursors
+Total number of precursors identified based on different confidence metrics.
+## [1] "Plot failed to render. Reason: Error: Upload ms1_extracted.tsv\n"Missing Data, Precursor Level
+Plotting the Jaccard Index for identified precursors for all channel combinations.
+## [1] "Plot failed to render. Reason: Error in `[.data.frame`(data()[[\"report\"]], , c(\"Raw.file\", \"Ms1.Area\", : undefined columns selected\n"Missing Data, Protein Level
+Plotting the Jaccard Index for identified precursors for all channel combinations.
+## [1] "Plot failed to render. Reason: Error in `[.data.frame`(data()[[\"report\"]], , c(\"Raw.file\", \"Ms1.Area\", : undefined columns selected\n"Feature Detection
+Features Identified by Charge
+Identified features are reported based on the charge. Precursors quantified in seperate channels are treated as separate precursors..
+## [1] "Plot failed to render. Reason: Error: Please provide a features.tsv file\n"Isotopes Identified per Feature
+The number of isotopes identified is shown for features detected in the Dinosaur search.
+## [1] "Plot failed to render. Reason: Error: Please provide a features.tsv file\n"Number of Scans per feature
+The number of MS1 scans is shown for all features identified in the Dinosaur search.
+## [1] "Plot failed to render. Reason: Error: Please provide a features.tsv file\n"Retention Length of Features at Base
+Plotting the retention length of identified features at the base.
+## [1] "Plot failed to render. Reason: Error: Please provide a features.tsv file\n"Features Identified across Gradient
+The frequency of precursor identifications based on the Dinosaur search is plotted across the chromatographic gradient.
+## [1] "Plot failed to render. Reason: Error: Please provide a features.tsv file\n"Features Identified across m/z
+The frequency of precursor identifications based on the Dinosaur search is plotted across the mass to charge ratio.
+## [1] "Plot failed to render. Reason: Error: Please provide a features.tsv file\n"Feature Intensity Distribution
+The distribution of integrated intensities is shown for identified features.
+## [1] "Plot failed to render. Reason: Error: Please provide a features.tsv file\n"