Contributing to CHARLIE¶
Proposing changes with issues¶
If you want to make a change, it's a good idea to first open an issue and make sure someone from the team agrees that it’s needed.
If you've decided to work on an issue, assign yourself to the issue so others will know you're working on it.
Pull request process¶
We use GitHub Flow as our collaboration process. Follow the steps below for detailed instructions on contributing changes to CHARLIE.
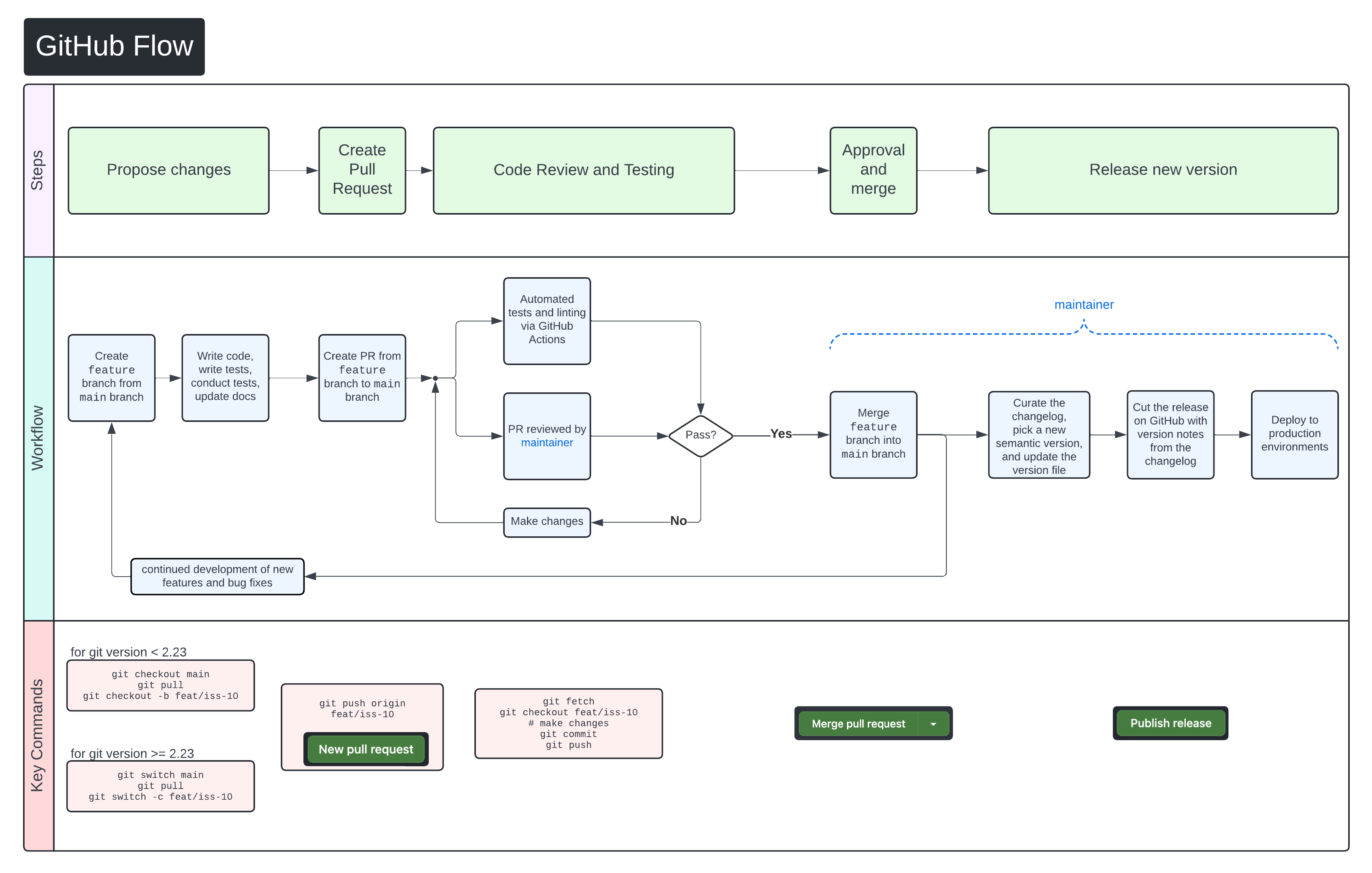
Clone the repo¶
If you are a member of CCBR, you can clone this repository to your computer or development environment. Otherwise, you will first need to fork the repo and clone your fork. You only need to do this step once.
git clone https://github.com/CCBR/CHARLIE
+ How to contribute - CHARLIE (Circrnas in Host And viRuses anaLysis pIpEline) Contributing to CHARLIE¶
Proposing changes with issues¶
If you want to make a change, it's a good idea to first open an issue and make sure someone from the team agrees that it’s needed.
If you've decided to work on an issue, assign yourself to the issue so others will know you're working on it.
Pull request process¶
We use GitHub Flow as our collaboration process. Follow the steps below for detailed instructions on contributing changes to CHARLIE.
Clone the repo¶
If you are a member of CCBR, you can clone this repository to your computer or development environment. Otherwise, you will first need to fork the repo and clone your fork. You only need to do this step once.
git clone https://github.com/CCBR/CHARLIE
Cloning into 'CHARLIE'...
remote: Enumerating objects: 1136, done.
remote: Counting objects: 100% (463/463), done.
remote: Compressing objects: 100% (357/357), done.
remote: Total 1136 (delta 149), reused 332 (delta 103), pack-reused 673
Receiving objects: 100% (1136/1136), 11.01 MiB | 9.76 MiB/s, done.
Resolving deltas: 100% (530/530), done.
cd CHARLIE
If this is your first time cloning the repo, you may need to install dependencies¶
-
Install snakemake and singularity or docker if needed (biowulf already has these available as modules).
-
Install the python dependencies with pip
pip install .
If you're developing on biowulf, you can use our shared conda environment which already has these dependencies installed
. "/data/CCBR_Pipeliner/db/PipeDB/Conda/etc/profile.d/conda.sh"
diff --git a/dev/flowchart/index.html b/dev/flowchart/index.html
index a2a48f0..f5a117c 100644
--- a/dev/flowchart/index.html
+++ b/dev/flowchart/index.html
@@ -1 +1 @@
- Workflow - CHARLIE (Circrnas in Host And viRuses anaLysis pIpEline) CHARLIE workflow¶

DISCLAIMER: This chart is for v0.8.x may be slightly outdated.
\ No newline at end of file
+ Workflow - CHARLIE (Circrnas in Host And viRuses anaLysis pIpEline) CHARLIE workflow¶
DISCLAIMER: This chart is for v0.8.x may be slightly outdated.
\ No newline at end of file
diff --git a/dev/index.html b/dev/index.html
index ed4bdb2..9c47f57 100644
--- a/dev/index.html
+++ b/dev/index.html
@@ -1,4 +1,5 @@
- CHARLIE (Circrnas in Host And viRuses anaLysis pIpEline) CHARLIE¶
Circrnas in Host And viRuses anaLysis pIpEline








See the website for detailed information, documentation, and examples: https://ccbr.github.io/CHARLIE/
Table of Contents¶
- CHARLIE
- Table of Contents
- 1. Introduction
- 2. Flowchart
- 3. Software Dependencies
- 4. Usage
- 5. License
- 6. Testing
1. Introduction¶
Circrnas in Host And viRuses anaLysis pIpEline
Things to know about CHARLIE:
- Snakemake workflow to detect, annotate and quantify (DAQ) host and viral circular RNAs.
- Primirarily developed to run on BIOWULF
- Reach out to Vishal Koparde for questions/comments/requests.
This circularRNA detection pipeline uses CIRCExplorer2, CIRI2 and many other tools in parallel to detect, quantify and annotate circRNAs. Here is a list of tools that can be run using CHARLIE:
circRNA Detection Tool Aligner(s) Run by default CIRCExplorer2 STAR1 Yes CIRI2 BWA1 Yes CIRCExplorer2 BWA1 Yes CLEAR STAR1 Yes DCC STAR2 Yes circRNAFinder STAR3 Yes find_circ Bowtie2 Yes MapSplice BWA2 No NCLScan NovoAlign No
Note: STAR1, STAR2, STAR3 denote 3 different sets of alignment parameters, etc.
Note: BWA1, BWA2 denote 2 different alignment parameters, etc.
2. Flowchart¶
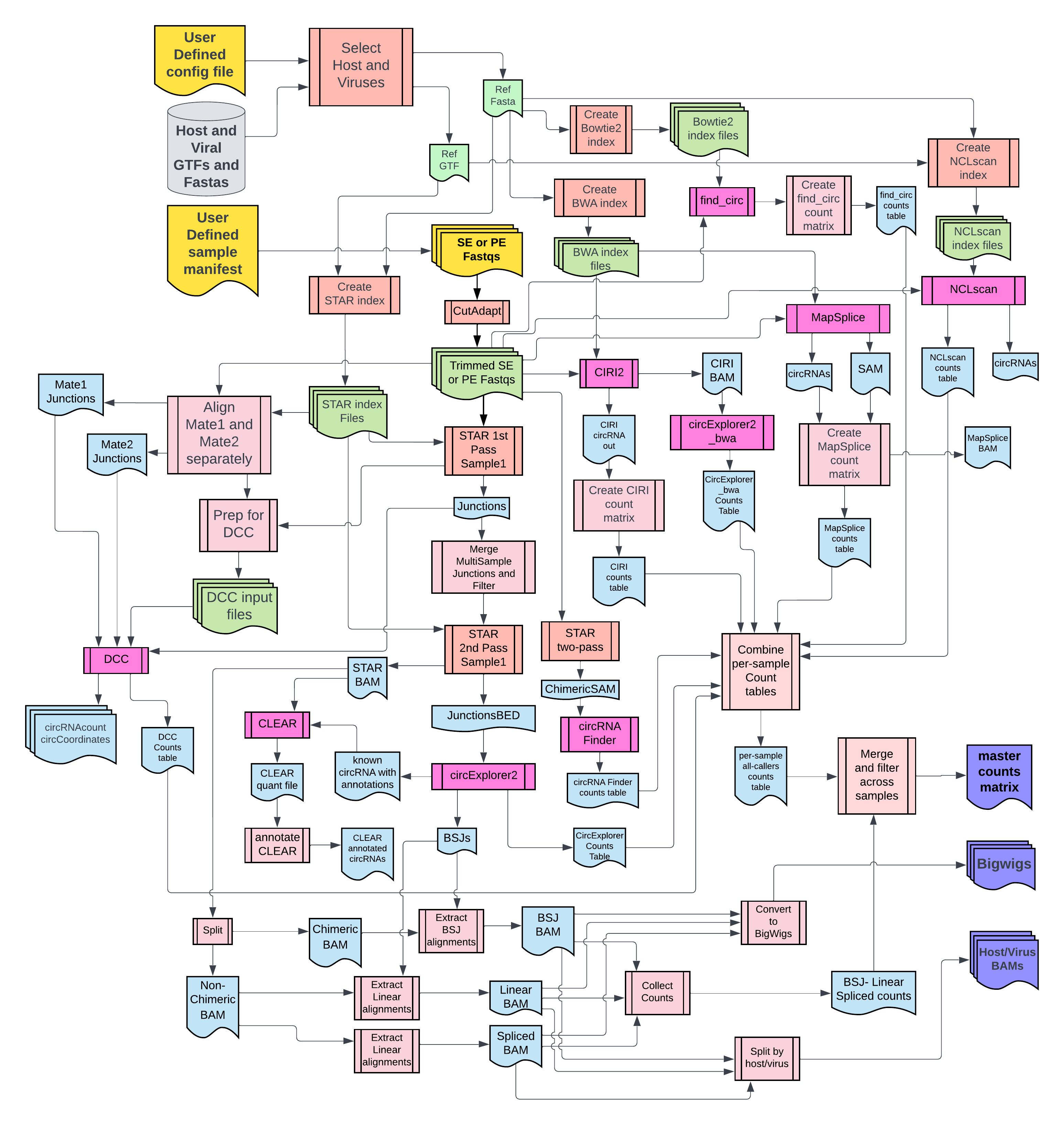
For complete documentation, view the website https://CCBR.github.io/CHARLIE/.
⚠️ DISCLAIMER: New circRNA tools have been added CHARLIE and the documentation is currently out of date!
3. Software Dependencies¶
The following version of various bioinformatics tools are using within CHARLIE:
tool version blat 3.5 bedtools 2.30.0 bowtie 2-2.5.1 bowtie 1.3.1 bwa 0.7.17 circexplorer2 2.3.8 cufflinks 2.2.1 cutadapt 4.4 fastqc 0.11.9 hisat 2.2.2.1 java 18.0.1.1 multiqc 1.9 parallel 20231122 perl 5.34 picard 2.27.3 python 2.7 python 3.8 sambamba 0.8.2 samtools 1.16.1 STAR 2.7.6a stringtie 2.2.1 ucsc 450 R 4.0.5 novocraft 4.03.05
4. Usage¶
% ./charlie
+ CHARLIE (Circrnas in Host And viRuses anaLysis pIpEline) CHARLIE¶
Circrnas in Host And viRuses anaLysis pIpEline
See the website for detailed information, documentation, and examples: https://ccbr.github.io/CHARLIE/
Table of Contents¶
- CHARLIE
- Table of Contents
- 1. Introduction
- 2. Flowchart
- 3. Software Dependencies
- 4. Usage
- 5. License
- 6. Testing
1. Introduction¶
Circrnas in Host And viRuses anaLysis pIpEline
Things to know about CHARLIE:
- Snakemake workflow to detect, annotate and quantify (DAQ) host and viral circular RNAs.
- Primirarily developed to run on BIOWULF
- Reach out to Vishal Koparde for questions/comments/requests.
This circularRNA detection pipeline uses CIRCExplorer2, CIRI2 and many other tools in parallel to detect, quantify and annotate circRNAs. Here is a list of tools that can be run using CHARLIE:
circRNA Detection Tool Aligner(s) Run by default CIRCExplorer2 STAR1 Yes CIRI2 BWA1 Yes CIRCExplorer2 BWA1 Yes CLEAR STAR1 Yes DCC STAR2 Yes circRNAFinder STAR3 Yes find_circ Bowtie2 Yes MapSplice BWA2 No NCLScan NovoAlign No
Note: STAR1, STAR2, STAR3 denote 3 different sets of alignment parameters, etc.
Note: BWA1, BWA2 denote 2 different alignment parameters, etc.
2. Flowchart¶
For complete documentation, view the website https://CCBR.github.io/CHARLIE/.
⚠️ DISCLAIMER: New circRNA tools have been added CHARLIE and the documentation is currently out of date!
3. Software Dependencies¶
CHARLIE is already installed on biowulf. It is included in the ccbrpipeliner module from release 7 onward. To load the module run:
module load ccbrpipeliner/7
+
The following version of various bioinformatics tools are using within CHARLIE:
tool version blat 3.5 bedtools 2.30.0 bowtie 2-2.5.1 bowtie 1.3.1 bwa 0.7.17 circexplorer2 2.3.8 cufflinks 2.2.1 cutadapt 4.4 fastqc 0.11.9 hisat 2.2.2.1 java 18.0.1.1 multiqc 1.9 parallel 20231122 perl 5.34 picard 2.27.3 python 2.7 python 3.8 sambamba 0.8.2 samtools 1.16.1 STAR 2.7.6a stringtie 2.2.1 ucsc 450 R 4.0.5 novocraft 4.03.05
4. Usage¶
charlie
##########################################################################################
@@ -49,7 +50,7 @@
##########################################################################################
USAGE:
- bash /data/Ziegelbauer_lab/Pipelines/circRNA/activeDev/charlie -w/--workdir=<WORKDIR> -m/--runmode=<RUNMODE>
+ charlie -w/--workdir=<WORKDIR> -m/--runmode=<RUNMODE>
Required Arguments:
1. WORKDIR : [Type: String]: Absolute or relative path to the output folder with write permissions.
@@ -78,21 +79,21 @@
Example commands:
- bash /data/Ziegelbauer_lab/Pipelines/circRNA/activeDev/charlie -w=/my/output/folder -m=init
- bash /data/Ziegelbauer_lab/Pipelines/circRNA/activeDev/charlie -w=/my/output/folder -m=dryrun
- bash /data/Ziegelbauer_lab/Pipelines/circRNA/activeDev/charlie -w=/my/output/folder -m=run
+ charlie -w=/my/output/folder -m=init
+ charlie -w=/my/output/folder -m=dryrun
+ charlie -w=/my/output/folder -m=run
##########################################################################################
VersionInfo:
- python : 3.7
- snakemake : 7.19.1
- pipeline_home : /vf/users/Ziegelbauer_lab/Pipelines/circRNA/activeDev
- git commit/tag : 1ae5ca091976364369784f67adffbbbf1dcdb7d5 v0.8-197-g1ae5ca0
+ python : 3
+ snakemake : 7
+ pipeline_home : /gpfs/gsfs10/users/CCBR_Pipeliner/Pipelines/CHARLIE/.v0.11.1
+ git commit/tag : 613fb617f1ed426fb8900f98e599ca0497a67cc0 v0.11.0-49-g613fb61
##########################################################################################
5. License¶
MIT License
Copyright © 2021 Vishal Koparde
Permission is hereby granted, free of charge, to any person obtaining a copy of this software and associated documentation files (the "Software"), to deal in the Software without restriction, including without limitation the rights to use, copy, modify, merge, publish, distribute, sublicense, and/or sell copies of the Software, and to permit persons to whom the Software is furnished to do so, subject to the following conditions:
The above copyright notice and this permission notice shall be included in all copies or substantial portions of the Software.
THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
6. Testing¶
Init¶
Run init mode:
bash <path to charlie> -w=<path to output dir> -m=init
-
This will create the folder provided by -w=. The user should have write permission to this folder.
Dry-run¶
Test data (1 paired-end subsample and 1 single-end subsample) have been including under the .tests/dummy_fastqs folder. After running in -m=init, samples.tsv should be edited to point the copies of the above mentioned samples with the column headers:
- sampleName
- path_to_R1_fastq
- path_to_R2_fastq
Column path_to_R2_fastq will be blank in case of single-end samples.
After editing samples.tsv, dry run should be run:
bash <path to charlie> -w=<path to output dir> -m=dryrun
+
This will create the folder provided by -w=. The user should have write permission to this folder.
Dry-run¶
Test data (1 paired-end subsample and 1 single-end subsample) have been including under the /data/CCBR_Pipeliner/testdata/circRNA/human folder. After running in -m=init, samples.tsv should be edited to point the copies of the above mentioned samples with the column headers:
- sampleName
- path_to_R1_fastq
- path_to_R2_fastq
Column path_to_R2_fastq will be blank in case of single-end samples.
After editing samples.tsv, dry run should be run:
bash <path to charlie> -w=<path to output dir> -m=dryrun
This will create the reference fasta and gtf file based on the selections made in the config.yaml.
Run¶
If -m=dryrun was successful, then simply do -m=run. The output will look something like this
... ... skipping ~1000 lines
...
...
@@ -140,9 +141,10 @@
This was a dry-run (flag -n). The order of jobs does not reflect the order of execution.
Running...
14743440
-
6.1 Test Data¶
The .tests/dummy_fastqs folder in the repo has test dataset:
% tree .tests/dummy_fastqs
-.tests/dummy_fastqs
-├── GI1_N.R1.fastq.gz
-├── GI1_N.R2.fastq.gz
-└── GI1_T.R1.fastq.gz
+
6.1 Test Data¶
The /data/CCBR_Pipeliner/testdata/circRNA/human folder in the repo has test dataset:
tree /data/CCBR_Pipeliner/testdata/circRNA/human
+/data/CCBR_Pipeliner/testdata/circRNA/human
+├── GI1_N_ss.R1.fastq.gz
+├── GI1_N_ss.R2.fastq.gz
+├── GI1_T_ss.R1.fastq.gz
+└── samples.tsv
GI1_N is a PE sample while GI1_T is a SE sample.
6.2 Expected Output¶
Expected output from the sample data is stored under .tests/expected_output.
More details about running test data can be found here.
DISCLAIMER:
CHARLIE is built to be run only on BIOWULF. A newer HPC-agnostic version of CHARLIE is planned for 2024.
\ No newline at end of file
diff --git a/dev/platforms/index.html b/dev/platforms/index.html
index 482197e..d320fcd 100644
--- a/dev/platforms/index.html
+++ b/dev/platforms/index.html
@@ -1,3 +1,3 @@
- Platforms - CHARLIE (Circrnas in Host And viRuses anaLysis pIpEline) Platforms
CHARLIE was originally developed to run on biowulf, but it can run on other computing platforms too. There are a few additional steps to configure CHARLIE.
TODO
- Clone CHARLIE.
git clone https://github.com/CCBR/charlie
+ Platforms - CHARLIE (Circrnas in Host And viRuses anaLysis pIpEline) Platforms
CHARLIE was originally developed to run on biowulf, but it can run on other computing platforms too. There are a few additional steps to configure CHARLIE.
TODO
- Clone CHARLIE.
git clone https://github.com/CCBR/charlie
- Initialize your project working directory.
-
Create a directory of reference files.
-
Edit your project's config file.
-
If you are using a SLURM job scheduler, edit cluster.json and submit_script.sbatch.
\ No newline at end of file
diff --git a/dev/references/index.html b/dev/references/index.html
index 0187b5d..162b4a0 100644
--- a/dev/references/index.html
+++ b/dev/references/index.html
@@ -1,4 +1,4 @@
- References - CHARLIE (Circrnas in Host And viRuses anaLysis pIpEline) References
References¶
The reference sequences comprises of the host genome and the viral genomes.
Fasta¶
hg38 and mm39 genome builds are chosen to represent hosts. Ribosomal sequences (45S, 5S) are downloaded from NCBI. hg38 and mm39 were masked for rRNA sequence and 45S and 5S sequences from NCBI are appended as separate chromosomes. The following viral sequences were appended to the rRNA masked hg38 reference:
HOSTS:
+ References - CHARLIE (Circrnas in Host And viRuses anaLysis pIpEline) References
References¶
The reference sequences comprises of the host genome and the viral genomes.
Fasta¶
hg38 and mm39 genome builds are chosen to represent hosts. Ribosomal sequences (45S, 5S) are downloaded from NCBI. hg38 and mm39 were masked for rRNA sequence and 45S and 5S sequences from NCBI are appended as separate chromosomes. The following viral sequences were appended to the rRNA masked hg38 reference:
HOSTS:
* hg38 [Human]
* mm39 [Mouse]
diff --git a/dev/search/search_index.json b/dev/search/search_index.json
index 0872d24..2dcd306 100644
--- a/dev/search/search_index.json
+++ b/dev/search/search_index.json
@@ -1 +1 @@
-{"config":{"lang":["en"],"separator":"[\\s\\-]+","pipeline":["stopWordFilter"]},"docs":[{"location":"","title":"CHARLIE","text":"Circrnas in Host And viRuses anaLysis pIpEline
See the website for detailed information, documentation, and examples: https://ccbr.github.io/CHARLIE/
"},{"location":"#table-of-contents","title":"Table of Contents","text":" - CHARLIE
- Table of Contents
- 1. Introduction
- 2. Flowchart
- 3. Software Dependencies
- 4. Usage
- 5. License
- 6. Testing
- 6.1 Test data
- 6.2 Expected output
"},{"location":"#1-introduction","title":"1. Introduction","text":"Circrnas in Host And viRuses anaLysis pIpEline
Things to know about CHARLIE:
- Snakemake workflow to detect, annotate and quantify (DAQ) host and viral circular RNAs.
- Primirarily developed to run on BIOWULF
- Reach out to Vishal Koparde for questions/comments/requests.
This circularRNA detection pipeline uses CIRCExplorer2, CIRI2 and many other tools in parallel to detect, quantify and annotate circRNAs. Here is a list of tools that can be run using CHARLIE:
circRNA Detection Tool Aligner(s) Run by default CIRCExplorer2 STAR1 Yes CIRI2 BWA1 Yes CIRCExplorer2 BWA1 Yes CLEAR STAR1 Yes DCC STAR2 Yes circRNAFinder STAR3 Yes find_circ Bowtie2 Yes MapSplice BWA2 No NCLScan NovoAlign No Note: STAR1, STAR2, STAR3 denote 3 different sets of alignment parameters, etc.
Note: BWA1, BWA2 denote 2 different alignment parameters, etc.
"},{"location":"#2-flowchart","title":"2. Flowchart","text":"For complete documentation, view the website https://CCBR.github.io/CHARLIE/.
\u26a0\ufe0f DISCLAIMER: New circRNA tools have been added CHARLIE and the documentation is currently out of date!
"},{"location":"#3-software-dependencies","title":"3. Software Dependencies","text":"The following version of various bioinformatics tools are using within CHARLIE:
tool version blat 3.5 bedtools 2.30.0 bowtie 2-2.5.1 bowtie 1.3.1 bwa 0.7.17 circexplorer2 2.3.8 cufflinks 2.2.1 cutadapt 4.4 fastqc 0.11.9 hisat 2.2.2.1 java 18.0.1.1 multiqc 1.9 parallel 20231122 perl 5.34 picard 2.27.3 python 2.7 python 3.8 sambamba 0.8.2 samtools 1.16.1 STAR 2.7.6a stringtie 2.2.1 ucsc 450 R 4.0.5 novocraft 4.03.05"},{"location":"#4-usage","title":"4. Usage","text":" % ./charlie\n\n\n##########################################################################################\n\nWelcome to\n _______ __ __ _______ ______ ___ ___ _______\n| || | | || _ || _ | | | | | | |\n| || |_| || |_| || | || | | | | | ___|\n| || || || |_||_ | | | | | |___\n| _|| || || __ || |___ | | | ___|\n| |_ | _ || _ || | | || || | | |___\n|_______||__| |__||__| |__||___| |_||_______||___| |_______|\n\nC_ircrnas in H_ost A_nd vi_R_uses ana_L_ysis p_I_p_E_line\n\n##########################################################################################\n\nThis pipeline was built by CCBR (https://bioinformatics.ccr.cancer.gov/ccbr)\nPlease contact Vishal Koparde for comments/questions (vishal.koparde@nih.gov)\n\n##########################################################################################\n\nCHARLIE can be used to DAQ(Detect/Annotate/Quantify) circRNAs in hosts and viruses.\n\nHere is the list of hosts and viruses that are currently supported:\n\nHOSTS:\n * hg38 [Human]\n * mm39 [Mouse]\n\nADDITIVES:\n * ERCC [External RNA Control Consortium sequences]\n * BAC16Insert [insert from rKSHV.219-derived BAC clone of the full-length KSHV genome]\n\nVIRUSES:\n * NC_007605.1 [Human gammaherpesvirus 4 (Epstein-Barr virus)]\n * NC_006273.2 [Human betaherpesvirus 5 (Cytomegalovirus )]\n * NC_001664.4 [Human betaherpesvirus 6A (HHV-6A)]\n * NC_000898.1 [Human betaherpesvirus 6B (HHV-6B)]\n * NC_001716.2 [Human betaherpesvirus 7 (HHV-7)]\n * NC_009333.1 [Human gammaherpesvirus 8 (KSHV)]\n * NC_045512.2 [Severe acute respiratory syndrome(SARS)-related coronavirus]\n * MN485971.1 [HIV from Belgium]\n * NC_001806.2 [Human alphaherpesvirus 1 (Herpes simplex virus type 1)](strain 17) (HSV-1)]\n * KT899744.1 [HSV-1 strain KOS]\n * MH636806.1 [MHV68 (Murine herpesvirus 68 strain WUMS)]\n\n##########################################################################################\n\nUSAGE:\n bash /data/Ziegelbauer_lab/Pipelines/circRNA/activeDev/charlie -w/--workdir=<WORKDIR> -m/--runmode=<RUNMODE>\n\nRequired Arguments:\n1. WORKDIR : [Type: String]: Absolute or relative path to the output folder with write permissions.\n\n2. RUNMODE : [Type: String] Valid options:\n * init : initialize workdir\n * dryrun : dry run snakemake to generate DAG\n * run : run with slurm\n * runlocal : run without submitting to sbatch\n ADVANCED RUNMODES (use with caution!!)\n * unlock : unlock WORKDIR if locked by snakemake NEVER UNLOCK WORKDIR WHERE PIPELINE IS CURRENTLY RUNNING!\n * reconfig : recreate config file in WORKDIR (debugging option) EDITS TO config.yaml WILL BE LOST!\n * reset : DELETE workdir dir and re-init it (debugging option) EDITS TO ALL FILES IN WORKDIR WILL BE LOST!\n * printbinds: print singularity binds (paths)\n * local : same as runlocal\n\nOptional Arguments:\n\n--singcache|-c : singularity cache directory. Default is `/data/${USER}/.singularity` if available, or falls back to `${WORKDIR}/.singularity`. Use this flag to specify a different singularity cache directory.\n--host|-g : supply host at command line. hg38 or mm39. (--runmode=init only)\n--additives|-a : supply comma-separated list of additives at command line. ERCC or BAC16Insert or both (--runmode=init only)\n--viruses|-v : supply comma-separated list of viruses at command line (--runmode=init only)\n--manifest|-s : absolute path to samples.tsv. This will be copied to output folder (--runmode=init only)\n--changegrp|-z : change group to \"Ziegelbauer_lab\" before running anything. Biowulf-only. Useful for correctly setting permissions.\n--help|-h : print this help\n\n\nExample commands:\n bash /data/Ziegelbauer_lab/Pipelines/circRNA/activeDev/charlie -w=/my/output/folder -m=init\n bash /data/Ziegelbauer_lab/Pipelines/circRNA/activeDev/charlie -w=/my/output/folder -m=dryrun\n bash /data/Ziegelbauer_lab/Pipelines/circRNA/activeDev/charlie -w=/my/output/folder -m=run\n\n##########################################################################################\n\nVersionInfo:\n python : 3.7\n snakemake : 7.19.1\n pipeline_home : /vf/users/Ziegelbauer_lab/Pipelines/circRNA/activeDev\n git commit/tag : 1ae5ca091976364369784f67adffbbbf1dcdb7d5 v0.8-197-g1ae5ca0\n\n##########################################################################################\n
"},{"location":"#5-license","title":"5. License","text":"MIT License
Copyright \u00a9 2021 Vishal Koparde
Permission is hereby granted, free of charge, to any person obtaining a copy of this software and associated documentation files (the \"Software\"), to deal in the Software without restriction, including without limitation the rights to use, copy, modify, merge, publish, distribute, sublicense, and/or sell copies of the Software, and to permit persons to whom the Software is furnished to do so, subject to the following conditions:
The above copyright notice and this permission notice shall be included in all copies or substantial portions of the Software.
THE SOFTWARE IS PROVIDED \"AS IS\", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
"},{"location":"#6-testing","title":"6. Testing","text":""},{"location":"#init","title":"Init","text":"Run init mode:
bash <path to charlie> -w=<path to output dir> -m=init\n
This will create the folder provided by -w=. The user should have write permission to this folder.
"},{"location":"#dry-run","title":"Dry-run","text":"Test data (1 paired-end subsample and 1 single-end subsample) have been including under the .tests/dummy_fastqs folder. After running in -m=init, samples.tsv should be edited to point the copies of the above mentioned samples with the column headers:
- sampleName
- path_to_R1_fastq
- path_to_R2_fastq
Column path_to_R2_fastq will be blank in case of single-end samples.
After editing samples.tsv, dry run should be run:
bash <path to charlie> -w=<path to output dir> -m=dryrun\n
This will create the reference fasta and gtf file based on the selections made in the config.yaml.
"},{"location":"#run","title":"Run","text":"If -m=dryrun was successful, then simply do -m=run. The output will look something like this
... ... skipping ~1000 lines\n...\n...\nJob stats:\njob count min threads max threads\n--------------------------------------------- ------- ----------\nall 1 1 1\nannotate_clear_output 2 1 1\ncircExplorer 2 2 2\ncircExplorer_bwa 2 2 2\ncircrnafinder 2 1 1\nciri 2 56 56\nclear 2 2 2\ncreate_bowtie2_index 1 1 1\ncreate_bwa_index 1 1 1\ncreate_circExplorer_BSJ_bam 2 4 4\ncreate_circExplorer_linear_spliced_bams 2 56 56\ncreate_circExplorer_merged_found_counts_table 2 1 1\ncreate_hq_bams 2 1 1\ncreate_index 1 56 56\ncreate_master_counts_file 1 1 1\ncutadapt 2 56 56\ndcc 2 4 4\ndcc_create_samplesheets 2 1 1\nestimate_duplication 2 1 1\nfastqc 2 4 4\nfind_circ 2 56 56\nfind_circ_align 2 56 56\nmerge_SJ_tabs 1 2 2\nmerge_alignment_stats 1 1 1\nmerge_genecounts 1 1 1\nmerge_per_sample 2 1 1\nstar1p 2 56 56\nstar2p 2 56 56\nstar_circrnafinder 2 56 56\ntotal 52 1 56\n\nReasons:\n (check individual jobs above for details)\n input files updated by another job:\n alignment_stats, all, annotate_clear_output, circExplorer, circExplorer_bwa, circrnafinder, ciri, clear, create_circExplorer_BSJ_bam, create_circExplorer_linear_spliced_bams, create_circExplorer_merged_found_counts_table, create_hq_bams, create_master_counts_file, dcc, dcc_create_samplesheets, estimate_duplication, fastqc, find_circ, find_circ_align, merge_SJ_tabs, merge_alignment_stats, merge_genecounts, merge_per_sample, star1p, star2p, star_circrnafinder\n missing output files:\n alignment_stats, annotate_clear_output, circExplorer, circExplorer_bwa, circrnafinder, ciri, clear, create_bowtie2_index, create_bwa_index, create_circExplorer_BSJ_bam, create_circExplorer_linear_spliced_bams, create_circExplorer_merged_found_counts_table, create_hq_bams, create_index, create_master_counts_file, cutadapt, dcc, dcc_create_samplesheets, estimate_duplication, fastqc, find_circ, find_circ_align, merge_SJ_tabs, merge_alignment_stats, merge_genecounts, merge_per_sample, star1p, star2p, star_circrnafinder\n\nThis was a dry-run (flag -n). The order of jobs does not reflect the order of execution.\nRunning...\n14743440\n
"},{"location":"#61-test-data","title":"6.1 Test Data","text":"The .tests/dummy_fastqs folder in the repo has test dataset:
% tree .tests/dummy_fastqs\n.tests/dummy_fastqs\n\u251c\u2500\u2500 GI1_N.R1.fastq.gz\n\u251c\u2500\u2500 GI1_N.R2.fastq.gz\n\u2514\u2500\u2500 GI1_T.R1.fastq.gz\n
GI1_N is a PE sample while GI1_T is a SE sample.
"},{"location":"#62-expected-output","title":"6.2 Expected Output","text":"Expected output from the sample data is stored under .tests/expected_output.
More details about running test data can be found here.
DISCLAIMER:
CHARLIE is built to be run only on BIOWULF. A newer HPC-agnostic version of CHARLIE is planned for 2024.
"},{"location":"CHANGELOG/","title":"Changelog","text":""},{"location":"CHANGELOG/#charlie-development-version","title":"CHARLIE development version","text":""},{"location":"CHANGELOG/#bug-fixes","title":"bug fixes","text":" - CHARLIE was falsely throwing a file permissions error for tempdir values containing bash variables. (#118, @kelly-sovacool)
- Singularity bind paths were not being set properly. (#119, @kelly-sovacool)
- Update docker containers to set
$PYTHONPATH. (#119, #125, @kelly-sovacool) - Otherwise, this environment variable can be carried over and cause package conflicts when singularity is not run with
-C. - Also use
python -E to ensure the $PYTHONPATH is not carried over. (#129, @kelly-sovacool) - Fix
reconfig to correctly replace variables in the config file. (#121, @kelly-sovacool) - Prevent using excessive memory when copying reference files. (#126, @kelly-sovacool)
- Fix missing output files due to file system latency and use real (absolute) paths where possible. (#130, @kelly-sovacool)
"},{"location":"CHANGELOG/#charlie-0110","title":"CHARLIE 0.11.0","text":" - Major updates to convert CHARLIE from a biowulf-specific to a platform-agnostic pipeline (#102, @kelly-sovacool):
- All rules now use containers instead of envmodules.
- Default config and cluster config files are provided for use on biowulf and FRCE.
- New entry
TEMPDIR in the config file sets the temporary directory location for rules that require transient storage. - New
--singcache argument to provide a singularity cache dir location. The singularity cache dir is automatically set inside /data/$USER/ or $WORKDIR/ if --singcache is not provided. - Minor documentation improvements. (#114, @kelly-sovacool)
"},{"location":"CHANGELOG/#charlie-0101","title":"CHARLIE 0.10.1","text":" - strand are reported together, strand from all callers are reported,
- both + and - flanking sites are reported,
- rev-comp function updated,
- updated versions of tools to match available tools on BIOWULF.
"},{"location":"CHANGELOG/#charlie-090","title":"CHARLIE 0.9.0","text":"Significant upgrades since the last release:
- updates to wrapper script, many new arguments/options added
- new per-sample counts table format
- new all-sample master counts matrix with min-nreads filtering and ntools column to show number of tools supporting the circRNA call
- new version of Snakemake
- cluster_status script added for forced completion of pipeline upon TIMEOUTs
- updated flowchart from lucid charts
- added circRNAfinder, find_circ, circExplorer2_bwa and other tools
- optimized execution and resource requirements
- updated viral annotations (Thanks Sara!)
- new method to extract linear counts, create linear BAMs using circExplorer2 outputs
- new job reporting using jobby and its derivatives
- separated creation of BWA and BOWTIE2 index from creation of STAR index to speed things up
- parallelized find_circ
- better cleanup (eg. deleting _STARgenome folders, etc.) for much smaller digital footprint
- multitude of comments throughout the snakefiles including listing of output file column descriptions
- preliminary GH actions added
"},{"location":"CHANGELOG/#charlie-070","title":"CHARLIE 0.7.0","text":" - 5 circRNA callers
- all-sample counts matrix with annotations
"},{"location":"CHANGELOG/#charlie-069","title":"CHARLIE 0.6.9","text":" - Optimized pysam scripts
- fixed premature completion of singularity rules
"},{"location":"CHANGELOG/#charlie-065","title":"CHARLIE 0.6.5","text":" - updated config.yaml to use the latest HSV-1 annotations received from Sarah (050421)
"},{"location":"CHANGELOG/#charlie-064","title":"CHARLIE 0.6.4","text":" - create linear reads BAM file
- create linear reads BigWigs for each region in the .regions file.
"},{"location":"CHANGELOG/#charlie-063","title":"CHARLIE 0.6.3","text":" - QOS not working for Taka... removed from cluster.json
- recall rule requires python/3.7 ... env module updated
"},{"location":"CHANGELOG/#charlie-062","title":"CHARLIE 0.6.2","text":" - BSJ files are in BSJ subfolder... bug fix for v0.6.1
"},{"location":"CHANGELOG/#charlie-061","title":"CHARLIE 0.6.1","text":" - customBSJs recalled from STAR alignments
- only for PE
- removes erroneously called CircExplorer BSJs
- create sense and anti-sense BSJ BAMs and BW for each reference (host+viruses)
- find reads which contribute to CIRI BSJs but not on the STAR list of BSJ reads, see if they contribute to novel (not called by STAR) BSJs and append novel BSJs to customBSJ list
"},{"location":"CHANGELOG/#charlie-060","title":"CHARLIE 0.6.0","text":"cutadapt_min_length to cutadapt rule... setting it to 15 in config (for miRNAs, Biot and short viral features)
"},{"location":"CHANGELOG/#charlie-050","title":"CHARLIE 0.5.0","text":" run_clear is now set to True (as default)circ_quant replaces clear_quant in the CLEAR rule. In order words, we are reusing the STAR alignment file and the circExplorer2 output file for running CLEAR. No need to run HISAT2 and TopHat (fusion-search with Bowtie1). This is much quicker.- Using picard to estimate duplicates using MarkDuplicates
- Generating a per-run multiqc HTML report
- Using eulerr R package to generate CIRI-CircExplorer circRNA Venn diagrams and include them in the mulitqc report
- Gather per job cluster metadata like queue time, run time, job state etc. Stats are compiled in HPC_summary file
- CLEAR pipeline quant.txt file is annotated for known circRNAs
WORKDIR can now be a relative path- bam2bw conversion fix for BSJ and spliced_reads. Issue closed!
"},{"location":"CHANGELOG/#charlie-040","title":"CHARLIE 0.4.0","text":" - CLEAR added.
- wrapper script (
run_circrna_daq.sh) added for local and cluster execution. - \"spliced reads only\" bam created and split by regions
"},{"location":"CHANGELOG/#charlie-030","title":"CHARLIE 0.3.0","text":" - Lookup table for hg38 to hg19 circRNA annotations is updated... this eliminate one-to-many hits from the previous version
- BSJs extracted as different bam file.
- flowchart added
- adding slurmjobid to log/err file names
- v0.3.1 has significant (>10X) performance improvements at BSJ bam creation
- v0.3.3 splits BSJ bams into human and viral bams, and also converts them to bigwigs
- v0.3.4 adds hg38_rRNA_masked_plus_rRNA_plus_viruses_plus_ERCC reference (source:Sarah)
"},{"location":"CHANGELOG/#charlie-020","title":"CHARLIE 0.2.0","text":" - SE support added .. PE/SE samples handled concurrently
envmodules used in Snakemake in place of module load statements
"},{"location":"CHANGELOG/#charlie-010","title":"CHARLIE 0.1.0","text":" - base version
- PE only support
"},{"location":"contributing/","title":"Contributing to CHARLIE","text":""},{"location":"contributing/#proposing-changes-with-issues","title":"Proposing changes with issues","text":"If you want to make a change, it's a good idea to first open an issue and make sure someone from the team agrees that it\u2019s needed.
If you've decided to work on an issue, assign yourself to the issue so others will know you're working on it.
"},{"location":"contributing/#pull-request-process","title":"Pull request process","text":"We use GitHub Flow as our collaboration process. Follow the steps below for detailed instructions on contributing changes to CHARLIE.
"},{"location":"contributing/#clone-the-repo","title":"Clone the repo","text":"If you are a member of CCBR, you can clone this repository to your computer or development environment. Otherwise, you will first need to fork the repo and clone your fork. You only need to do this step once.
git clone https://github.com/CCBR/CHARLIE\n
Cloning into 'CHARLIE'... remote: Enumerating objects: 1136, done. remote: Counting objects: 100% (463/463), done. remote: Compressing objects: 100% (357/357), done. remote: Total 1136 (delta 149), reused 332 (delta 103), pack-reused 673 Receiving objects: 100% (1136/1136), 11.01 MiB | 9.76 MiB/s, done. Resolving deltas: 100% (530/530), done.
cd CHARLIE\n
"},{"location":"contributing/#if-this-is-your-first-time-cloning-the-repo-you-may-need-to-install-dependencies","title":"If this is your first time cloning the repo, you may need to install dependencies","text":" -
Install snakemake and singularity or docker if needed (biowulf already has these available as modules).
-
Install the python dependencies with pip
pip install .\n
If you're developing on biowulf, you can use our shared conda environment which already has these dependencies installed
. \"/data/CCBR_Pipeliner/db/PipeDB/Conda/etc/profile.d/conda.sh\"\nconda activate py311\n
- Install
pre-commit if you don't already have it. Then from the repo's root directory, run
pre-commit install\n
This will install the repo's pre-commit hooks. You'll only need to do this step the first time you clone the repo.
"},{"location":"contributing/#create-a-branch","title":"Create a branch","text":"Create a Git branch for your pull request (PR). Give the branch a descriptive name for the changes you will make, such as iss-10 if it is for a specific issue.
# create a new branch and switch to it\ngit branch iss-10\ngit switch iss-10\n
Switched to a new branch 'iss-10'
"},{"location":"contributing/#make-your-changes","title":"Make your changes","text":"Edit the code, write and run tests, and update the documentation as needed.
"},{"location":"contributing/#test","title":"test","text":"Changes to the python package code will also need unit tests to demonstrate that the changes work as intended. We write unit tests with pytest and store them in the tests/ subdirectory. Run the tests with python -m pytest.
If you change the workflow, please run the workflow with the test profile and make sure your new feature or bug fix works as intended.
"},{"location":"contributing/#document","title":"document","text":"If you have added a new feature or changed the API of an existing feature, you will likely need to update the documentation in docs/.
"},{"location":"contributing/#commit-and-push-your-changes","title":"Commit and push your changes","text":"If you're not sure how often you should commit or what your commits should consist of, we recommend following the \"atomic commits\" principle where each commit contains one new feature, fix, or task. Learn more about atomic commits here: https://www.freshconsulting.com/insights/blog/atomic-commits/
First, add the files that you changed to the staging area:
git add path/to/changed/files/\n
Then make the commit. Your commit message should follow the Conventional Commits specification. Briefly, each commit should start with one of the approved types such as feat, fix, docs, etc. followed by a description of the commit. Take a look at the Conventional Commits specification for more detailed information about how to write commit messages.
git commit -m 'feat: create function for awesome feature'\n
pre-commit will enforce that your commit message and the code changes are styled correctly and will attempt to make corrections if needed.
Check for added large files..............................................Passed Fix End of Files.........................................................Passed Trim Trailing Whitespace.................................................Failed
- hook id: trailing-whitespace
- exit code: 1
- files were modified by this hook > Fixing path/to/changed/files/file.txt > codespell................................................................Passed style-files..........................................(no files to check)Skipped readme-rmd-rendered..................................(no files to check)Skipped use-tidy-description.................................(no files to check)Skipped
In the example above, one of the hooks modified a file in the proposed commit, so the pre-commit check failed. You can run git diff to see the changes that pre-commit made and git status to see which files were modified. To proceed with the commit, re-add the modified file(s) and re-run the commit command:
git add path/to/changed/files/file.txt\ngit commit -m 'feat: create function for awesome feature'\n
This time, all the hooks either passed or were skipped (e.g. hooks that only run on R code will not run if no R files were committed). When the pre-commit check is successful, the usual commit success message will appear after the pre-commit messages showing that the commit was created.
Check for added large files..............................................Passed Fix End of Files.........................................................Passed Trim Trailing Whitespace.................................................Passed codespell................................................................Passed style-files..........................................(no files to check)Skipped readme-rmd-rendered..................................(no files to check)Skipped use-tidy-description.................................(no files to check)Skipped Conventional Commit......................................................Passed > [iss-10 9ff256e] feat: create function for awesome feature 1 file changed, 22 insertions(+), 3 deletions(-)
Finally, push your changes to GitHub:
git push\n
If this is the first time you are pushing this branch, you may have to explicitly set the upstream branch:
git push --set-upstream origin iss-10\n
Enumerating objects: 7, done. Counting objects: 100% (7/7), done. Delta compression using up to 10 threads Compressing objects: 100% (4/4), done. Writing objects: 100% (4/4), 648 bytes | 648.00 KiB/s, done. Total 4 (delta 3), reused 0 (delta 0), pack-reused 0 remote: Resolving deltas: 100% (3/3), completed with 3 local objects. remote: remote: Create a pull request for 'iss-10' on GitHub by visiting: remote: https://github.com/CCBR/CHARLIE/pull/new/iss-10 remote: To https://github.com/CCBR/CHARLIE > > [new branch] iss-10 -> iss-10 branch 'iss-10' set up to track 'origin/iss-10'.
We recommend pushing your commits often so they will be backed up on GitHub. You can view the files in your branch on GitHub at https://github.com/CCBR/CHARLIE/tree/<your-branch-name> (replace <your-branch-name> with the actual name of your branch).
"},{"location":"contributing/#create-the-pr","title":"Create the PR","text":"Once your branch is ready, create a PR on GitHub: https://github.com/CCBR/CHARLIE/pull/new/
Select the branch you just pushed:
Edit the PR title and description. The title should briefly describe the change. Follow the comments in the template to fill out the body of the PR, and you can delete the comments (everything between <!-- and -->) as you go. Be sure to fill out the checklist, checking off items as you complete them or striking through any irrelevant items. When you're ready, click 'Create pull request' to open it.
Optionally, you can mark the PR as a draft if you're not yet ready for it to be reviewed, then change it later when you're ready.
"},{"location":"contributing/#wait-for-a-maintainer-to-review-your-pr","title":"Wait for a maintainer to review your PR","text":"We will do our best to follow the tidyverse code review principles: https://code-review.tidyverse.org/. The reviewer may suggest that you make changes before accepting your PR in order to improve the code quality or style. If that's the case, continue to make changes in your branch and push them to GitHub, and they will appear in the PR.
Once the PR is approved, the maintainer will merge it and the issue(s) the PR links will close automatically. Congratulations and thank you for your contribution!
"},{"location":"contributing/#after-your-pr-has-been-merged","title":"After your PR has been merged","text":"After your PR has been merged, update your local clone of the repo by switching to the main branch and pulling the latest changes:
git checkout main\ngit pull\n
It's a good idea to run git pull before creating a new branch so it will start from the most recent commits in main.
"},{"location":"contributing/#helpful-links-for-more-information","title":"Helpful links for more information","text":" - GitHub Flow
- semantic versioning guidelines
- changelog guidelines
- tidyverse code review principles
- reproducible examples
- nf-core extensions for VS Code
"},{"location":"flowchart/","title":"CHARLIE workflow","text":"DISCLAIMER: This chart is for v0.8.x may be slightly outdated.
"},{"location":"platforms/","title":"Platforms","text":"CHARLIE was originally developed to run on biowulf, but it can run on other computing platforms too. There are a few additional steps to configure CHARLIE.
TODO
- Clone CHARLIE.
git clone https://github.com/CCBR/charlie\n
- Initialize your project working directory.
\n
-
Create a directory of reference files.
-
Edit your project's config file.
-
If you are using a SLURM job scheduler, edit cluster.json and submit_script.sbatch.
"},{"location":"references/","title":"References","text":""},{"location":"references/#references","title":"References","text":"The reference sequences comprises of the host genome and the viral genomes.
"},{"location":"references/#fasta","title":"Fasta","text":"hg38 and mm39 genome builds are chosen to represent hosts. Ribosomal sequences (45S, 5S) are downloaded from NCBI. hg38 and mm39 were masked for rRNA sequence and 45S and 5S sequences from NCBI are appended as separate chromosomes. The following viral sequences were appended to the rRNA masked hg38 reference:
HOSTS:\n * hg38 [Human]\n * mm39 [Mouse]\n\nADDITIVES:\n * ERCC [External RNA Control Consortium sequences]\n * BAC16Insert [insert from rKSHV.219-derived BAC clone of the full-length KSHV genome]\n\nVIRUSES:\n * NC_007605.1 [Human gammaherpesvirus 4 (Epstein-Barr virus)]\n * NC_006273.2 [Human betaherpesvirus 5 (Cytomegalovirus )]\n * NC_001664.4 [Human betaherpesvirus 6A (HHV-6A)]\n * NC_000898.1 [Human betaherpesvirus 6B (HHV-6B)]\n * NC_001716.2 [Human betaherpesvirus 7 (HHV-7)]\n * NC_009333.1 [Human gammaherpesvirus 8 (KSHV)]\n * NC_045512.2 [Severe acute respiratory syndrome(SARS)-related coronavirus]\n * MN485971.1 [HIV from Belgium]\n * NC_001806.2 [Human alphaherpesvirus 1 (Herpes simplex virus type 1)](strain 17) (HSV-1)]\n * KT899744.1 [HSV-1 strain KOS]\n * MH636806.1 [MHV68 (Murine herpesvirus 68 strain WUMS)]\n
Location: The entire resource bundle is available at /data/CCBR_Pipeliner/db/PipeDB/charlie/fastas_gtfs on BIOWULF. This location also have additional bash scripts required for aggregating annotations and building indices required by different aligners.
When -m=dryrun is run for the first time after initialization (-m=init), the appropriate host+additives+viruses fasta and gtf files are created on the fly, which are then used to build aligner reference indexes automatically.
"},{"location":"tutorial/","title":"Tutorial","text":""},{"location":"tutorial/#prerequisites","title":"Prerequisites","text":" -
Biowulf account: Biowulf account can be requested here.
-
Membership to Ziegelbauer user group on Biowulf. You can check this by typing the following command:
% groups\n
output:
CCBR kopardevn Ziegelbauer_lab\n
If Ziegelbauer_lab is not listed then you can email a request to be added to the groups here
"},{"location":"tutorial/#location","title":"Location","text":"Different versions of circRNA DAQ pipeline have been parked at /data/Ziegelbauer_lab/Pipelines/circRNA
% ls /data/Ziegelbauer_lab/Pipelines/circRNA\n
output:
v0.1.0\nv0.10.0\nv0.10.0-dev\nv0.2.1\nv0.3.3\nv0.4.2\nv0.5.2\nv0.6.5\nv0.7.0\nv0.8\nv0.9.0\n
The exacts versions listed here may changed as newer versions are added. Also, the dev version is pointing to the most recent untagged version of the pipeline (use at own risk!)
"},{"location":"tutorial/#init","title":"Init","text":"To get help about the pipeline you can run:
% bash /data/Ziegelbauer_lab/Pipelines/circRNA/v0.10.0-dev/charlie\n
output:
##########################################################################################\n\nWelcome to charlie(v0.10.0-dev)\n _______ __ __ _______ ______ ___ ___ _______\n| || | | || _ || _ | | | | | | |\n| || |_| || |_| || | || | | | | | ___|\n| || || || |_||_ | | | | | |___\n| _|| || || __ || |___ | | | ___|\n| |_ | _ || _ || | | || || | | |___\n|_______||__| |__||__| |__||___| |_||_______||___| |_______|\n\nC_ircrnas in H_ost A_nd vi_R_uses ana_L_ysis p_I_p_E_line\n\n##########################################################################################\n\nThis pipeline was built by CCBR (https://bioinformatics.ccr.cancer.gov/ccbr)\nPlease contact Vishal Koparde for comments/questions (vishal.koparde@nih.gov)\n\n##########################################################################################\n\nCHARLIE can be used to DAQ(Detect/Annotate/Quantify) circRNAs in hosts and viruses.\n\nHere is the list of hosts and viruses that are currently supported:\n\nHOSTS:\n * hg38 [Human]\n * mm39 [Mouse]\n\nADDITIVES:\n * ERCC [External RNA Control Consortium sequences]\n * BAC16Insert [insert from rKSHV.219-derived BAC clone of the full-length KSHV genome]\n\nVIRUSES:\n * NC_007605.1 [Human gammaherpesvirus 4 (Epstein-Barr virus)]\n * NC_006273.2 [Human betaherpesvirus 5 (Cytomegalovirus )]\n * NC_001664.4 [Human betaherpesvirus 6A (HHV-6A)]\n * NC_000898.1 [Human betaherpesvirus 6B (HHV-6B)]\n * NC_001716.2 [Human betaherpesvirus 7 (HHV-7)]\n * NC_009333.1 [Human gammaherpesvirus 8 (KSHV)]\n * NC_045512.2 [Severe acute respiratory syndrome(SARS)-related coronavirus]\n * MN485971.1 [HIV from Belgium]\n * NC_001806.2 [Human alphaherpesvirus 1 (Herpes simplex virus type 1)](strain 17) (HSV-1)]\n * KT899744.1 [HSV-1 strain KOS]\n * MH636806.1 [MHV68 (Murine herpesvirus 68 strain WUMS)]\n\n##########################################################################################\n\nUSAGE:\n bash /data/Ziegelbauer_lab/Pipelines/circRNA/v0.10.0-dev/charlie -w/--workdir=<WORKDIR> -m/--runmode=<RUNMODE>\n\nRequired Arguments:\n1. WORKDIR : [Type: String]: Absolute or relative path to the output folder with write permissions.\n\n2. RUNMODE : [Type: String] Valid options:\n * init : initialize workdir\n * dryrun : dry run snakemake to generate DAG\n * run : run with slurm\n * runlocal : run without submitting to sbatch\n ADVANCED RUNMODES (use with caution!!)\n * unlock : unlock WORKDIR if locked by snakemake NEVER UNLOCK WORKDIR WHERE PIPELINE IS CURRENTLY RUNNING!\n * reconfig : recreate config file in WORKDIR (debugging option) EDITS TO config.yaml WILL BE LOST!\n * reset : DELETE workdir dir and re-init it (debugging option) EDITS TO ALL FILES IN WORKDIR WILL BE LOST!\n * printbinds: print singularity binds (paths)\n * local : same as runlocal\n\nOptional Arguments:\n\n--singcache|-c : singularity cache directory. Default is `/data/${USER}/.singularity` if available, or falls back to `${WORKDIR}/.singularity`. Use this flag to specify a different singularity cache directory.\n--host|-g : supply host at command line. hg38 or mm39. (--runmode=init only)\n--additives|-a : supply comma-separated list of additives at command line. ERCC or BAC16Insert or both (--runmode=init only)\n--viruses|-v : supply comma-separated list of viruses at command line (--runmode=init only)\n--manifest|-s : absolute path to samples.tsv. This will be copied to output folder (--runmode=init only)\n--changegrp|-z : change group to \"Ziegelbauer_lab\" before running anything. Biowulf-only. Useful for correctly setting permissions.\n--help|-h : print this help\n\n\nExample commands:\n bash /data/Ziegelbauer_lab/Pipelines/circRNA/v0.10.0-dev/charlie -w=/my/output/folder -m=init\n bash /data/Ziegelbauer_lab/Pipelines/circRNA/v0.10.0-dev/charlie -w=/my/output/folder -m=dryrun\n bash /data/Ziegelbauer_lab/Pipelines/circRNA/v0.10.0-dev/charlie -w=/my/output/folder -m=run\n\n##########################################################################################\n\nVersionInfo:\n python : 3.7\n snakemake : 7.19.1\n pipeline_home : /vf/users/Ziegelbauer_lab/Pipelines/circRNA/v0.10.0-dev\n git commit/tag : b2cf2f089788651041b16bf4378c2c5172c13cb2 v0.10.0-2-gb2cf2f0\n\n##########################################################################################\n
NOTE: You can replace v0.10.0 in the above command with the latest version to use a newer version. run_circrna_daq.sh was called test.sh in versions older than v0.4.0.
To initial the working directory run:
% bash <path to charlie> -w=<path to output dir> -m=init\n
This assumes that <path to output dir> does not exist before running the above command and is at a location where write permissions are available.
The above command creates <path to output dir> folder and creates 2 subfolders logs and stats inside that folder along with config.yaml and samples.tsv files.
% tree <path to output dir>\n
"},{"location":"tutorial/#configyaml","title":"config.yaml","text":"This file is used to fine tune the execution of the pipeline by setting:
- sample sheet location ... aka
samples.tsv - the temporary directory -- make sure this is correct for your computing environment.
- which circRNA finding tools to use by editing these:
- run_clear: True
- run_dcc: True
- run_mapsplice: False
- run_circRNAFinder: True
- run_nclscan: False
- run_findcirc: False
- describes the location of other resources/indexes/tools etc. Generally, these do NOT need to be changed.
"},{"location":"tutorial/#samplestsv","title":"samples.tsv","text":"Tab delimited definition of sample sheet. The header is fixed and each row represents a sample. It has 3 columns:
- sampleName = Name of the sample. This has to be unique.
- path_to_R1_fastq = absolute path to the read1 fastq.gz file.
- path_to_R2_fastq = absolute path to the read2 fastq.gz file. If the sample was sequenced in single-end mode, then leave this blank.
The .tests/dummy_fastqs folder in the repo has test dataset:
% tree .tests/dummy_fastqs\n.tests/dummy_fastqs\n\u251c\u2500\u2500 GI1_N.R1.fastq.gz\n\u251c\u2500\u2500 GI1_N.R2.fastq.gz\n\u2514\u2500\u2500 GI1_T.R1.fastq.gz\n
GI1_N is a PE sample while GI1_T is a SE sample.
"},{"location":"tutorial/#dryrun","title":"Dryrun","text":"Once the samples.tsv file has been edited appropriately to include the desired samples, it is a good idea to dryrun the pipeline to ensure that everything will work as desired. Dryrun can be run as follows:
bash <path to charlie> -w=<path to output dir> -m=dryrun\n
This will create the reference fasta and gtf file based on the selections made in the config.yaml. Hence, can take a few minutes to run.
"},{"location":"tutorial/#run","title":"Run","text":"Upon verifying that dryrun is successful. You can then submit the job to the cluster using the following command:
bash <path to charlie> -w=<path to output dir> -m=run\n
which will produce something like this:
... ... skipping ~1000 lines\n...\n...\nJob stats:\njob count min threads max threads\n--------------------------------------------- ------- ----------\nall 1 1 1\nannotate_clear_output 2 1 1\ncircExplorer 2 2 2\ncircExplorer_bwa 2 2 2\ncircrnafinder 2 1 1\nciri 2 56 56\nclear 2 2 2\ncreate_bowtie2_index 1 1 1\ncreate_bwa_index 1 1 1\ncreate_circExplorer_BSJ_bam 2 4 4\ncreate_circExplorer_linear_spliced_bams 2 56 56\ncreate_circExplorer_merged_found_counts_table 2 1 1\ncreate_hq_bams 2 1 1\ncreate_index 1 56 56\ncreate_master_counts_file 1 1 1\ncutadapt 2 56 56\ndcc 2 4 4\ndcc_create_samplesheets 2 1 1\nestimate_duplication 2 1 1\nfastqc 2 4 4\nfind_circ 2 56 56\nfind_circ_align 2 56 56\nmerge_SJ_tabs 1 2 2\nmerge_alignment_stats 1 1 1\nmerge_genecounts 1 1 1\nmerge_per_sample 2 1 1\nstar1p 2 56 56\nstar2p 2 56 56\nstar_circrnafinder 2 56 56\ntotal 52 1 56\n\nReasons:\n (check individual jobs above for details)\n input files updated by another job:\n alignment_stats, all, annotate_clear_output, circExplorer, circExplorer_bwa, circrnafinder, ciri, clear, create_circExplorer_BSJ_bam, create_circExplorer_linear_spliced_bams, create_circExplorer_merged_found_counts_table, create_hq_bams, create_master_counts_file, dcc, dcc_create_samplesheets, estimate_duplication, fastqc, find_circ, find_circ_align, merge_SJ_tabs, merge_alignment_stats, merge_genecounts, merge_per_sample, star1p, star2p, star_circrnafinder\n missing output files:\n alignment_stats, annotate_clear_output, circExplorer, circExplorer_bwa, circrnafinder, ciri, clear, create_bowtie2_index, create_bwa_index, create_circExplorer_BSJ_bam, create_circExplorer_linear_spliced_bams, create_circExplorer_merged_found_counts_table, create_hq_bams, create_index, create_master_counts_file, cutadapt, dcc, dcc_create_samplesheets, estimate_duplication, fastqc, find_circ, find_circ_align, merge_SJ_tabs, merge_alignment_stats, merge_genecounts, merge_per_sample, star1p, star2p, star_circrnafinder\n\nThis was a dry-run (flag -n). The order of jobs does not reflect the order of execution.\nRunning...\n14743440\n
In this example, 14743440 is the jobid returned by the slurm job scheduler on biowulf. This means that the job was successfully submitted, it will spawn off other subjobs which in-turn will be run and outputs will be moved to the results folder created inside the working directory supplied at command line. You can check the status of your queue of jobs in biowulf running:
% squeue -u `whoami`\n
output:
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)\n 14743440 ccr,norm circRNA kopardev PD 0:00 1 (None)\n
ST in the above results stands for Status and PD means Pending. The status will change from pending(PD) to running(R) to completed as jobs are run on the cluster.
Next, just sit tight until the pipeline finishes. You can keep monitoring the queue as shown above. If there are no jobs running on biowulf, then your pipeline has finished (or errored out!)
Once completed the output should something like this:
% tree <path to output dir>\n
output:
.\n\u251c\u2500\u2500 cluster.json\n\u251c\u2500\u2500 config.yaml\n\u251c\u2500\u2500 dryrun.231222103505.log\n\u251c\u2500\u2500 fastqs\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R1.fastq.gz -> /data/CCBR_Pipeliner/testdata/circRNA/human/GI1_N_ss.R1.fastq.gz\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R2.fastq.gz -> /data/CCBR_Pipeliner/testdata/circRNA/human/GI1_N_ss.R2.fastq.gz\n\u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T.R1.fastq.gz -> /data/CCBR_Pipeliner/testdata/circRNA/human/GI1_T_ss.R1.fastq.gz\n\u251c\u2500\u2500 logs/snakemake.log.jobby.short\n\u251c\u2500\u2500 logs/snakemake.log.jobby.txt\n\u251c\u2500\u2500 logs\n\u2502 ... log files ...\n\u2502 ... skipping ...\n\u251c\u2500\u2500 nclscan.config\n\u251c\u2500\u2500 qc\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 fastqc\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R1_fastqc.html\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R1_fastqc.zip\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R1.trim_fastqc.html\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R1.trim_fastqc.zip\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R2_fastqc.html\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R2_fastqc.zip\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R2.trim_fastqc.html\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R2.trim_fastqc.zip\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.R1_fastqc.html\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.R1_fastqc.zip\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.R1.trim_fastqc.html\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T.R1.trim_fastqc.zip\n\u2502\u00a0\u00a0 \u2514\u2500\u2500 picard_MarkDuplicates\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.MarkDuplicates.metrics.txt\n\u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T.MarkDuplicates.metrics.txt\n\u251c\u2500\u2500 ref\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 bwa_index.log\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 gene_id_2_gene_name.tsv\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 NCLscan_index\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 AllRef.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 AllRef.fa.amb\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 AllRef.fa.ann\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 AllRef.fa.bwt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 AllRef.fa.pac\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 AllRef.fa.sa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 AllRef.ndx\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 RepChrM.fa\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.1.bt2\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.2.bt2\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.3.bt2\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.4.bt2\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.amb\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.ann\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.bwt\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.dummy.fa\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.fa\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.fa.byo_index\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.fa.fai\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.fa.regions\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.fa.regions.host\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.fa.regions.viruses\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.fa.sizes\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.fixed.gtf\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.genes.genepred\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.genes.genepred_w_geneid\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.gtf\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.pac\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.rev.1.bt2\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.rev.2.bt2\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.sa\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.transcripts.fa\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 separate_fastas\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr10.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr11.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr12.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr13.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr14.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr15.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr16.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr17.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr18.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr19.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr20.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr21.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr22.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr2.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr3.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr45S.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr4.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr5.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr5S.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr6.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr7.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr8.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr9.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chrM.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chrX.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chrY.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00002.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00003.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00004.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00009.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00012.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00013.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00014.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00016.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00017.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00019.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00022.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00024.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00025.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00028.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00031.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00033.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00034.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00035.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00039.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00040.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00041.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00042.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00043.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00044.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00046.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00048.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00051.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00053.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00054.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00057.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00058.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00059.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00060.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00061.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00062.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00067.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00069.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00071.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00073.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00074.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00075.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00076.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00077.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00078.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00079.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00081.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00083.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00084.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00085.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00086.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00092.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00095.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00096.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00097.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00098.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00099.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00104.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00108.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00109.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00111.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00112.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00113.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00116.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00117.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00120.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00123.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00126.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00130.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00131.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00134.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00136.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00137.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00138.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00142.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00143.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00144.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00145.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00147.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00148.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00150.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00154.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00156.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00157.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00158.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00160.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00162.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00163.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00164.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00165.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00168.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00170.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00171.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000008.2.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000009.2.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000194.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000195.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000205.2.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000208.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000213.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000214.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000216.2.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000218.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000219.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000220.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000221.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000224.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000225.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000226.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270302.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270303.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270304.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270305.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270310.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270311.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270312.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270315.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270316.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270317.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270320.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270322.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270329.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270330.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270333.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270334.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270335.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270336.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270337.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270338.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270340.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270362.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270363.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270364.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270366.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270371.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270372.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270373.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270374.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270375.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270376.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270378.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270379.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270381.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270382.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270383.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270384.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270385.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270386.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270387.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270388.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270389.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270390.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270391.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270392.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270393.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270394.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270395.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270396.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270411.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270412.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270414.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270417.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270418.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270419.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270420.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270422.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270423.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270424.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270425.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270429.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270435.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270438.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270442.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270448.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270465.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270466.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270467.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270468.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270507.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270508.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270509.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270510.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270511.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270512.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270515.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270516.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270517.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270518.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270519.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270521.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270522.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270528.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270529.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270530.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270538.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270539.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270544.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270548.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270579.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270580.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270581.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270582.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270583.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270584.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270587.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270588.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270589.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270590.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270591.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270593.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270706.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270707.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270708.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270709.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270710.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270711.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270712.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270713.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270714.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270715.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270716.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270717.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270718.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270719.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270720.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270721.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270722.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270723.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270724.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270725.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270726.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270727.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270728.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270729.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270730.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270731.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270732.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270733.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270734.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270735.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270736.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270737.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270738.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270739.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270740.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270741.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270742.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270743.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270744.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270745.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270746.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270747.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270748.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270749.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270750.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270751.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270752.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270753.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270754.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270755.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270756.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270757.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 NC_009333.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 separate_fastas.lst\n\u2502\u00a0\u00a0 \u2514\u2500\u2500 STAR_no_GTF\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 chrLength.txt\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 chrNameLength.txt\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 chrName.txt\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 chrStart.txt\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 Genome\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 genomeParameters.txt\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 Log.out\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 SA\n\u2502\u00a0\u00a0 \u2514\u2500\u2500 SAindex\n\u251c\u2500\u2500 results\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 alignmentstats.txt\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 circRNA_master_counts.tsv.gz\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 alignmentstats.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 circExplorer\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.back_spliced_junction.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.back_spliced_junction.strand_fixed.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.bam.bai\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.bed.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.foundcounts.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.minus.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.minus.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.minus.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.plus.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.plus.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.plus.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.circExplorer.annotation_counts.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.circExplorer.counts_table.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_circexplorer_parse.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.circRNA_known.filter1.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.circularRNA_known.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.hg38.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.hg38.BSJ.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.hg38.BSJ.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear_BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear_BSJ.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear_BSJ.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear_BSJ.hg38.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear_BSJ.hg38.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear_BSJ.NC_009333.1.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear_BSJ.NC_009333.1.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear.BSJ.readids.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear.hg38.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear.hg38.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear.NC_009333.1.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear.NC_009333.1.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear_spliced.counts.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.NC_009333.1.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.NC_009333.1.BSJ.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.NC_009333.1.BSJ.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.readcounts.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.rid2jid.tsv.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced_BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced_BSJ.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced_BSJ.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced_BSJ.hg38.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced_BSJ.hg38.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced_BSJ.NC_009333.1.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced_BSJ.NC_009333.1.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced.BSJ.readids.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced.hg38.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced.hg38.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced.NC_009333.1.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced.NC_009333.1.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 low_conf_GI1_N.circRNA_known.filter1.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 low_conf_GI1_N.circRNA_known.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 circExplorer_BWA\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.back_spliced_junction.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.back_spliced_junction.strand_fixed.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.circExplorer_bwa.annotation_counts.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_circexplorer_parse.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.circRNA_known.filter1.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.circularRNA_known.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 low_conf_GI1_N.circRNA_known.filter1.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 low_conf_GI1_N.circRNA_known.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 circRNA_finder\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.Chimeric.out.sorted.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.Chimeric.out.sorted.bam.bai\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.circRNA_finder.counts_table.tsv.filtered\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.filteredJunctions.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.s_filteredJunctions.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_N.s_filteredJunctions_fw.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ciri\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 CIRIerror.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.bwa.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.ciri.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.ciri.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.ciri.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.ciri.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_N.ciri.out.filtered\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 CLEAR\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 quant.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 quant.txt.annotated\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 DCC\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 CircCoordinates\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 CircRNACount\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 CircSkipJunctions\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 DCC-2023-12-20_1149.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.dcc.counts_table.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.dcc.counts_table.tsv.filtered\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 LinearCount\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 mate1.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 mate2.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 samplesheet.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 find_circ\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_anchors.fastq.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.unmapped.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_N.unmapped.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.circRNA_counts.txt.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 merge_per_sample.sh\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 STAR1p\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p1.Chimeric.out.junction\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p1.Log.final.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p1.Log.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p1.Log.progress.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p1.SJ.out.tab\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 mate1\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_mate1.Chimeric.out.junction\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_mate1.Log.final.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_mate1.Log.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_mate1.Log.progress.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_N_mate1.SJ.out.tab\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 mate2\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_mate2.Chimeric.out.junction\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_mate2.Chimeric.out.junction.fixed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_mate2.Log.final.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_mate2.Log.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_mate2.Log.progress.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_N_mate2.SJ.out.tab\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 STAR2p\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.chimeric.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.chimeric.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.Chimeric.out.junction\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.Log.final.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.Log.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.Log.progress.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.non_chimeric.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.non_chimeric.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.ReadsPerGene.out.tab\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_N_p2.SJ.out.tab\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 STAR_circRNAFinder\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.Chimeric.out.junction\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.Chimeric.out.sam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.Log.final.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.Log.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.Log.progress.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_N.SJ.out.tab\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 trim\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R1.trim.fastq.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_N.R2.trim.fastq.gz\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 alignmentstats.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 circExplorer\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.back_spliced_junction.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.back_spliced_junction.strand_fixed.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.bam.bai\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.bed.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.foundcounts.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.minus.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.minus.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.minus.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.plus.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.plus.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.plus.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.circExplorer.annotation_counts.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.circExplorer.counts_table.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_circexplorer_parse.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.circRNA_known.filter1.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.circularRNA_known.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.hg38.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.hg38.BSJ.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.hg38.BSJ.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear_BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear_BSJ.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear_BSJ.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear_BSJ.hg38.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear_BSJ.hg38.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear_BSJ.NC_009333.1.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear_BSJ.NC_009333.1.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear.BSJ.readids.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear.hg38.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear.hg38.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear.NC_009333.1.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear.NC_009333.1.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear_spliced.counts.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.NC_009333.1.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.NC_009333.1.BSJ.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.NC_009333.1.BSJ.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.readcounts.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.rid2jid.tsv.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced_BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced_BSJ.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced_BSJ.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced_BSJ.hg38.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced_BSJ.hg38.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced_BSJ.NC_009333.1.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced_BSJ.NC_009333.1.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced.BSJ.readids.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced.hg38.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced.hg38.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced.NC_009333.1.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced.NC_009333.1.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 low_conf_GI1_T.circRNA_known.filter1.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 low_conf_GI1_T.circRNA_known.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 circExplorer_BWA\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.back_spliced_junction.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.back_spliced_junction.strand_fixed.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.circExplorer_bwa.annotation_counts.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_circexplorer_parse.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.circRNA_known.filter1.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.circularRNA_known.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 low_conf_GI1_T.circRNA_known.filter1.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 low_conf_GI1_T.circRNA_known.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 circRNA_finder\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.Chimeric.out.sorted.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.Chimeric.out.sorted.bam.bai\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.circRNA_finder.counts_table.tsv.filtered\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.filteredJunctions.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.s_filteredJunctions.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T.s_filteredJunctions_fw.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ciri\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 CIRIerror.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.bwa.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.ciri.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.ciri.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.ciri.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.ciri.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T.ciri.out.filtered\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 CLEAR\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 quant.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 quant.txt.annotated\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 DCC\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 CircCoordinates\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 CircRNACount\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 CircSkipJunctions\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 DCC-2023-12-20_1206.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.dcc.counts_table.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.dcc.counts_table.tsv.filtered\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 LinearCount\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 mate1.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 mate2.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 samplesheet.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 find_circ\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_anchors.fastq.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.unmapped.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T.unmapped.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.circRNA_counts.txt.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 merge_per_sample.sh\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 STAR1p\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p1.Chimeric.out.junction\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p1.Chimeric.out.junction.circRNA\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p1.Chimeric.out.junction.circRNAmapped\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p1.Log.final.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p1.Log.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p1.Log.progress.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p1.SJ.out.tab\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 mate1\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T_mate1.Chimeric.out.junction\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 mate2\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T_mate2.Chimeric.out.junction\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 STAR2p\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.chimeric.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.chimeric.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.Chimeric.out.junction\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.Log.final.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.Log.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.Log.progress.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.non_chimeric.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.non_chimeric.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.ReadsPerGene.out.tab\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T_p2.SJ.out.tab\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 STAR_circRNAFinder\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.Chimeric.out.junction\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.Chimeric.out.sam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.Log.final.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.Log.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.Log.progress.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T.SJ.out.tab\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 trim\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.R1.trim.fastq.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T.R2.trim.fastq.gz\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 HQ_BSJ_bams\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.hg38.HQ_only.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.hg38.HQ_only.BSJ.bam.bai\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.HQ_only.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.HQ_only.BSJ.bam.bai\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.NC_009333.1.HQ_only.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.NC_009333.1.HQ_only.BSJ.bam.bai\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.hg38.HQ_only.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.hg38.HQ_only.BSJ.bam.bai\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.HQ_only.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.HQ_only.BSJ.bam.bai\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.NC_009333.1.HQ_only.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T.NC_009333.1.HQ_only.BSJ.bam.bai\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 pass1.out.tab\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 revstranded_STAR_GeneCounts.tsv\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 stranded_STAR_GeneCounts.tsv\n\u2502\u00a0\u00a0 \u2514\u2500\u2500 unstranded_STAR_GeneCounts.tsv\n\u251c\u2500\u2500 runinfo.yaml\n\u251c\u2500\u2500 runslurm_snakemake_report.html\n\u251c\u2500\u2500 samples.tsv\n\u251c\u2500\u2500 snakemake.log\n\u251c\u2500\u2500 snakemake.stats\n\u251c\u2500\u2500 stats\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 dryrun.231222102114.log\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 runinfo.modtime.yaml\n\u2502\u00a0\u00a0 \u2514\u2500\u2500 snakemake.20231222104214.log\n\u251c\u2500\u2500 submit_script.sbatch\n\u2514\u2500\u2500 tools.txt\n
"},{"location":"tutorial/#error-tracking","title":"Error tracking","text":"Using the following command to find FAILED jobs:
grep FAIL logs/snakemake.log.jobby.short\n
The above command also gives .err and .out log files which can give further insights on reasons for failure and changes required to be made for a successful run.
"},{"location":"tutorial/#expected-output","title":"Expected output:","text":"The main output file is results/circRNA_master_counts.tsv.gz. Here are the top 3 tiles from an example output:
Column_number Column_title Example_1 Example_2 Example_3 1 chrom GL000220.1 GL000220.1 GL000220.1 2 start 107635 112482 118578 3 end 151634 156427 118759 4 circExplorer_strand -1 -1 -1 5 circExplorer_bwa_strand . . . 6 ciri_strand -1 -1 -1 7 dcc_strand -1 -1 -1 8 circrnafinder_strand -1 -1 -1 9 flankingsites+ CC##GC GC##CC CC##GC 10 flankingsites- GG##GC GC##GG GG##GC 11 sample_name GI1_N GI1_N GI1_N 12 ntools 1 1 1 13 HQ N N N 14 circExplorer_read_count -1 -1 -1 15 circExplorer_found_BSJcounts -1 -1 -1 16 circExplorerfound_linear_BSJ+_counts -1 -1 -1 17 circExplorerfound_linear_spliced_BSJ+_counts -1 -1 -1 18 circExplorerfound_linear_BSJ-_counts -1 -1 -1 19 circExplorerfound_linear_spliced_BSJ-_counts -1 -1 -1 20 circExplorerfound_linear_BSJ._counts -1 -1 -1 21 circExplorerfound_linear_spliced_BSJ._counts -1 -1 -1 22 ciri_read_count -1 -1 -1 23 ciri_linear_read_count -1 -1 -1 24 circExplorer_bwa_read_count 3 7 3 25 dcc_read_count -1 -1 -1 26 dcc_linear_read_count -1 -1 -1 27 circrnafinder_read_count -1 -1 -1 28 hqcounts 1 1 1 29 nonhqcounts 0 0 0 30 circExplorer_annotation Unknown Unknown Unknown 31 ciri_annotation Unknown Unknown Unknown 32 circExplorer_bwa_annotation novel novel novel 33 dcc_gene Unknown Unknown Unknown 34 dcc_junction_type Unknown Unknown Unknown 35 dcc_annotation Unknown Unknown Unknown Expected output from the sample data is stored under .tests/expected_output.
"}]}
\ No newline at end of file
+{"config":{"lang":["en"],"separator":"[\\s\\-]+","pipeline":["stopWordFilter"]},"docs":[{"location":"","title":"CHARLIE","text":"Circrnas in Host And viRuses anaLysis pIpEline
See the website for detailed information, documentation, and examples: https://ccbr.github.io/CHARLIE/
"},{"location":"#table-of-contents","title":"Table of Contents","text":" - CHARLIE
- Table of Contents
- 1. Introduction
- 2. Flowchart
- 3. Software Dependencies
- 4. Usage
- 5. License
- 6. Testing
- 6.1 Test data
- 6.2 Expected output
"},{"location":"#1-introduction","title":"1. Introduction","text":"Circrnas in Host And viRuses anaLysis pIpEline
Things to know about CHARLIE:
- Snakemake workflow to detect, annotate and quantify (DAQ) host and viral circular RNAs.
- Primirarily developed to run on BIOWULF
- Reach out to Vishal Koparde for questions/comments/requests.
This circularRNA detection pipeline uses CIRCExplorer2, CIRI2 and many other tools in parallel to detect, quantify and annotate circRNAs. Here is a list of tools that can be run using CHARLIE:
circRNA Detection Tool Aligner(s) Run by default CIRCExplorer2 STAR1 Yes CIRI2 BWA1 Yes CIRCExplorer2 BWA1 Yes CLEAR STAR1 Yes DCC STAR2 Yes circRNAFinder STAR3 Yes find_circ Bowtie2 Yes MapSplice BWA2 No NCLScan NovoAlign No Note: STAR1, STAR2, STAR3 denote 3 different sets of alignment parameters, etc.
Note: BWA1, BWA2 denote 2 different alignment parameters, etc.
"},{"location":"#2-flowchart","title":"2. Flowchart","text":"For complete documentation, view the website https://CCBR.github.io/CHARLIE/.
\u26a0\ufe0f DISCLAIMER: New circRNA tools have been added CHARLIE and the documentation is currently out of date!
"},{"location":"#3-software-dependencies","title":"3. Software Dependencies","text":"CHARLIE is already installed on biowulf. It is included in the ccbrpipeliner module from release 7 onward. To load the module run:
module load ccbrpipeliner/7\n
The following version of various bioinformatics tools are using within CHARLIE:
tool version blat 3.5 bedtools 2.30.0 bowtie 2-2.5.1 bowtie 1.3.1 bwa 0.7.17 circexplorer2 2.3.8 cufflinks 2.2.1 cutadapt 4.4 fastqc 0.11.9 hisat 2.2.2.1 java 18.0.1.1 multiqc 1.9 parallel 20231122 perl 5.34 picard 2.27.3 python 2.7 python 3.8 sambamba 0.8.2 samtools 1.16.1 STAR 2.7.6a stringtie 2.2.1 ucsc 450 R 4.0.5 novocraft 4.03.05"},{"location":"#4-usage","title":"4. Usage","text":"charlie\n\n\n##########################################################################################\n\nWelcome to\n _______ __ __ _______ ______ ___ ___ _______\n| || | | || _ || _ | | | | | | |\n| || |_| || |_| || | || | | | | | ___|\n| || || || |_||_ | | | | | |___\n| _|| || || __ || |___ | | | ___|\n| |_ | _ || _ || | | || || | | |___\n|_______||__| |__||__| |__||___| |_||_______||___| |_______|\n\nC_ircrnas in H_ost A_nd vi_R_uses ana_L_ysis p_I_p_E_line\n\n##########################################################################################\n\nThis pipeline was built by CCBR (https://bioinformatics.ccr.cancer.gov/ccbr)\nPlease contact Vishal Koparde for comments/questions (vishal.koparde@nih.gov)\n\n##########################################################################################\n\nCHARLIE can be used to DAQ(Detect/Annotate/Quantify) circRNAs in hosts and viruses.\n\nHere is the list of hosts and viruses that are currently supported:\n\nHOSTS:\n * hg38 [Human]\n * mm39 [Mouse]\n\nADDITIVES:\n * ERCC [External RNA Control Consortium sequences]\n * BAC16Insert [insert from rKSHV.219-derived BAC clone of the full-length KSHV genome]\n\nVIRUSES:\n * NC_007605.1 [Human gammaherpesvirus 4 (Epstein-Barr virus)]\n * NC_006273.2 [Human betaherpesvirus 5 (Cytomegalovirus )]\n * NC_001664.4 [Human betaherpesvirus 6A (HHV-6A)]\n * NC_000898.1 [Human betaherpesvirus 6B (HHV-6B)]\n * NC_001716.2 [Human betaherpesvirus 7 (HHV-7)]\n * NC_009333.1 [Human gammaherpesvirus 8 (KSHV)]\n * NC_045512.2 [Severe acute respiratory syndrome(SARS)-related coronavirus]\n * MN485971.1 [HIV from Belgium]\n * NC_001806.2 [Human alphaherpesvirus 1 (Herpes simplex virus type 1)](strain 17) (HSV-1)]\n * KT899744.1 [HSV-1 strain KOS]\n * MH636806.1 [MHV68 (Murine herpesvirus 68 strain WUMS)]\n\n##########################################################################################\n\nUSAGE:\n charlie -w/--workdir=<WORKDIR> -m/--runmode=<RUNMODE>\n\nRequired Arguments:\n1. WORKDIR : [Type: String]: Absolute or relative path to the output folder with write permissions.\n\n2. RUNMODE : [Type: String] Valid options:\n * init : initialize workdir\n * dryrun : dry run snakemake to generate DAG\n * run : run with slurm\n * runlocal : run without submitting to sbatch\n ADVANCED RUNMODES (use with caution!!)\n * unlock : unlock WORKDIR if locked by snakemake NEVER UNLOCK WORKDIR WHERE PIPELINE IS CURRENTLY RUNNING!\n * reconfig : recreate config file in WORKDIR (debugging option) EDITS TO config.yaml WILL BE LOST!\n * reset : DELETE workdir dir and re-init it (debugging option) EDITS TO ALL FILES IN WORKDIR WILL BE LOST!\n * printbinds: print singularity binds (paths)\n * local : same as runlocal\n\nOptional Arguments:\n\n--singcache|-c : singularity cache directory. Default is `/data/${USER}/.singularity` if available, or falls back to `${WORKDIR}/.singularity`. Use this flag to specify a different singularity cache directory.\n--host|-g : supply host at command line. hg38 or mm39. (--runmode=init only)\n--additives|-a : supply comma-separated list of additives at command line. ERCC or BAC16Insert or both (--runmode=init only)\n--viruses|-v : supply comma-separated list of viruses at command line (--runmode=init only)\n--manifest|-s : absolute path to samples.tsv. This will be copied to output folder (--runmode=init only)\n--changegrp|-z : change group to \"Ziegelbauer_lab\" before running anything. Biowulf-only. Useful for correctly setting permissions.\n--help|-h : print this help\n\n\nExample commands:\n charlie -w=/my/output/folder -m=init\n charlie -w=/my/output/folder -m=dryrun\n charlie -w=/my/output/folder -m=run\n\n##########################################################################################\n\nVersionInfo:\n python : 3\n snakemake : 7\n pipeline_home : /gpfs/gsfs10/users/CCBR_Pipeliner/Pipelines/CHARLIE/.v0.11.1\n git commit/tag : 613fb617f1ed426fb8900f98e599ca0497a67cc0 v0.11.0-49-g613fb61\n\n##########################################################################################\n
"},{"location":"#5-license","title":"5. License","text":"MIT License
Copyright \u00a9 2021 Vishal Koparde
Permission is hereby granted, free of charge, to any person obtaining a copy of this software and associated documentation files (the \"Software\"), to deal in the Software without restriction, including without limitation the rights to use, copy, modify, merge, publish, distribute, sublicense, and/or sell copies of the Software, and to permit persons to whom the Software is furnished to do so, subject to the following conditions:
The above copyright notice and this permission notice shall be included in all copies or substantial portions of the Software.
THE SOFTWARE IS PROVIDED \"AS IS\", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
"},{"location":"#6-testing","title":"6. Testing","text":""},{"location":"#init","title":"Init","text":"Run init mode:
bash <path to charlie> -w=<path to output dir> -m=init\n
This will create the folder provided by -w=. The user should have write permission to this folder.
"},{"location":"#dry-run","title":"Dry-run","text":"Test data (1 paired-end subsample and 1 single-end subsample) have been including under the /data/CCBR_Pipeliner/testdata/circRNA/human folder. After running in -m=init, samples.tsv should be edited to point the copies of the above mentioned samples with the column headers:
- sampleName
- path_to_R1_fastq
- path_to_R2_fastq
Column path_to_R2_fastq will be blank in case of single-end samples.
After editing samples.tsv, dry run should be run:
bash <path to charlie> -w=<path to output dir> -m=dryrun\n
This will create the reference fasta and gtf file based on the selections made in the config.yaml.
"},{"location":"#run","title":"Run","text":"If -m=dryrun was successful, then simply do -m=run. The output will look something like this
... ... skipping ~1000 lines\n...\n...\nJob stats:\njob count min threads max threads\n--------------------------------------------- ------- ----------\nall 1 1 1\nannotate_clear_output 2 1 1\ncircExplorer 2 2 2\ncircExplorer_bwa 2 2 2\ncircrnafinder 2 1 1\nciri 2 56 56\nclear 2 2 2\ncreate_bowtie2_index 1 1 1\ncreate_bwa_index 1 1 1\ncreate_circExplorer_BSJ_bam 2 4 4\ncreate_circExplorer_linear_spliced_bams 2 56 56\ncreate_circExplorer_merged_found_counts_table 2 1 1\ncreate_hq_bams 2 1 1\ncreate_index 1 56 56\ncreate_master_counts_file 1 1 1\ncutadapt 2 56 56\ndcc 2 4 4\ndcc_create_samplesheets 2 1 1\nestimate_duplication 2 1 1\nfastqc 2 4 4\nfind_circ 2 56 56\nfind_circ_align 2 56 56\nmerge_SJ_tabs 1 2 2\nmerge_alignment_stats 1 1 1\nmerge_genecounts 1 1 1\nmerge_per_sample 2 1 1\nstar1p 2 56 56\nstar2p 2 56 56\nstar_circrnafinder 2 56 56\ntotal 52 1 56\n\nReasons:\n (check individual jobs above for details)\n input files updated by another job:\n alignment_stats, all, annotate_clear_output, circExplorer, circExplorer_bwa, circrnafinder, ciri, clear, create_circExplorer_BSJ_bam, create_circExplorer_linear_spliced_bams, create_circExplorer_merged_found_counts_table, create_hq_bams, create_master_counts_file, dcc, dcc_create_samplesheets, estimate_duplication, fastqc, find_circ, find_circ_align, merge_SJ_tabs, merge_alignment_stats, merge_genecounts, merge_per_sample, star1p, star2p, star_circrnafinder\n missing output files:\n alignment_stats, annotate_clear_output, circExplorer, circExplorer_bwa, circrnafinder, ciri, clear, create_bowtie2_index, create_bwa_index, create_circExplorer_BSJ_bam, create_circExplorer_linear_spliced_bams, create_circExplorer_merged_found_counts_table, create_hq_bams, create_index, create_master_counts_file, cutadapt, dcc, dcc_create_samplesheets, estimate_duplication, fastqc, find_circ, find_circ_align, merge_SJ_tabs, merge_alignment_stats, merge_genecounts, merge_per_sample, star1p, star2p, star_circrnafinder\n\nThis was a dry-run (flag -n). The order of jobs does not reflect the order of execution.\nRunning...\n14743440\n
"},{"location":"#61-test-data","title":"6.1 Test Data","text":"The /data/CCBR_Pipeliner/testdata/circRNA/human folder in the repo has test dataset:
tree /data/CCBR_Pipeliner/testdata/circRNA/human\n/data/CCBR_Pipeliner/testdata/circRNA/human\n\u251c\u2500\u2500 GI1_N_ss.R1.fastq.gz\n\u251c\u2500\u2500 GI1_N_ss.R2.fastq.gz\n\u251c\u2500\u2500 GI1_T_ss.R1.fastq.gz\n\u2514\u2500\u2500 samples.tsv\n
GI1_N is a PE sample while GI1_T is a SE sample.
"},{"location":"#62-expected-output","title":"6.2 Expected Output","text":"Expected output from the sample data is stored under .tests/expected_output.
More details about running test data can be found here.
DISCLAIMER:
CHARLIE is built to be run only on BIOWULF. A newer HPC-agnostic version of CHARLIE is planned for 2024.
"},{"location":"CHANGELOG/","title":"Changelog","text":""},{"location":"CHANGELOG/#charlie-development-version","title":"CHARLIE development version","text":" - CHARLIE was falsely throwing a file permissions error for tempdir values containing bash variables. (#118, @kelly-sovacool)
- Singularity bind paths were not being set properly. (#119, @kelly-sovacool)
- Update docker containers to set
$PYTHONPATH. (#119, #125, @kelly-sovacool) - Otherwise, this environment variable can be carried over and cause package conflicts when singularity is not run with
-C. - Also use
python -E to ensure the $PYTHONPATH is not carried over. (#129, @kelly-sovacool) - Fix
reconfig to correctly replace variables in the config file. (#121, @kelly-sovacool) - Prevent using excessive memory when copying reference files. (#126, @kelly-sovacool)
- Fix missing output files due to file system latency and use real (absolute) paths where possible. (#130, @kelly-sovacool)
- Update documentation to reflect biowulf usage and improved test dataset. (#132, @kelly-sovacool)
"},{"location":"CHANGELOG/#charlie-0110","title":"CHARLIE 0.11.0","text":" - Major updates to convert CHARLIE from a biowulf-specific to a platform-agnostic pipeline (#102, @kelly-sovacool):
- All rules now use containers instead of envmodules.
- Default config and cluster config files are provided for use on biowulf and FRCE.
- New entry
TEMPDIR in the config file sets the temporary directory location for rules that require transient storage. - New
--singcache argument to provide a singularity cache dir location. The singularity cache dir is automatically set inside /data/$USER/ or $WORKDIR/ if --singcache is not provided. - Minor documentation improvements. (#114, @kelly-sovacool)
"},{"location":"CHANGELOG/#charlie-0101","title":"CHARLIE 0.10.1","text":" - strand are reported together, strand from all callers are reported,
- both + and - flanking sites are reported,
- rev-comp function updated,
- updated versions of tools to match available tools on BIOWULF.
"},{"location":"CHANGELOG/#charlie-090","title":"CHARLIE 0.9.0","text":"Significant upgrades since the last release:
- updates to wrapper script, many new arguments/options added
- new per-sample counts table format
- new all-sample master counts matrix with min-nreads filtering and ntools column to show number of tools supporting the circRNA call
- new version of Snakemake
- cluster_status script added for forced completion of pipeline upon TIMEOUTs
- updated flowchart from lucid charts
- added circRNAfinder, find_circ, circExplorer2_bwa and other tools
- optimized execution and resource requirements
- updated viral annotations (Thanks Sara!)
- new method to extract linear counts, create linear BAMs using circExplorer2 outputs
- new job reporting using jobby and its derivatives
- separated creation of BWA and BOWTIE2 index from creation of STAR index to speed things up
- parallelized find_circ
- better cleanup (eg. deleting _STARgenome folders, etc.) for much smaller digital footprint
- multitude of comments throughout the snakefiles including listing of output file column descriptions
- preliminary GH actions added
"},{"location":"CHANGELOG/#charlie-070","title":"CHARLIE 0.7.0","text":" - 5 circRNA callers
- all-sample counts matrix with annotations
"},{"location":"CHANGELOG/#charlie-069","title":"CHARLIE 0.6.9","text":" - Optimized pysam scripts
- fixed premature completion of singularity rules
"},{"location":"CHANGELOG/#charlie-065","title":"CHARLIE 0.6.5","text":" - updated config.yaml to use the latest HSV-1 annotations received from Sarah (050421)
"},{"location":"CHANGELOG/#charlie-064","title":"CHARLIE 0.6.4","text":" - create linear reads BAM file
- create linear reads BigWigs for each region in the .regions file.
"},{"location":"CHANGELOG/#charlie-063","title":"CHARLIE 0.6.3","text":" - QOS not working for Taka... removed from cluster.json
- recall rule requires python/3.7 ... env module updated
"},{"location":"CHANGELOG/#charlie-062","title":"CHARLIE 0.6.2","text":" - BSJ files are in BSJ subfolder... bug fix for v0.6.1
"},{"location":"CHANGELOG/#charlie-061","title":"CHARLIE 0.6.1","text":" - customBSJs recalled from STAR alignments
- only for PE
- removes erroneously called CircExplorer BSJs
- create sense and anti-sense BSJ BAMs and BW for each reference (host+viruses)
- find reads which contribute to CIRI BSJs but not on the STAR list of BSJ reads, see if they contribute to novel (not called by STAR) BSJs and append novel BSJs to customBSJ list
"},{"location":"CHANGELOG/#charlie-060","title":"CHARLIE 0.6.0","text":"cutadapt_min_length to cutadapt rule... setting it to 15 in config (for miRNAs, Biot and short viral features)
"},{"location":"CHANGELOG/#charlie-050","title":"CHARLIE 0.5.0","text":" run_clear is now set to True (as default)circ_quant replaces clear_quant in the CLEAR rule. In order words, we are reusing the STAR alignment file and the circExplorer2 output file for running CLEAR. No need to run HISAT2 and TopHat (fusion-search with Bowtie1). This is much quicker.- Using picard to estimate duplicates using MarkDuplicates
- Generating a per-run multiqc HTML report
- Using eulerr R package to generate CIRI-CircExplorer circRNA Venn diagrams and include them in the mulitqc report
- Gather per job cluster metadata like queue time, run time, job state etc. Stats are compiled in HPC_summary file
- CLEAR pipeline quant.txt file is annotated for known circRNAs
WORKDIR can now be a relative path- bam2bw conversion fix for BSJ and spliced_reads. Issue closed!
"},{"location":"CHANGELOG/#charlie-040","title":"CHARLIE 0.4.0","text":" - CLEAR added.
- wrapper script (
run_circrna_daq.sh) added for local and cluster execution. - \"spliced reads only\" bam created and split by regions
"},{"location":"CHANGELOG/#charlie-030","title":"CHARLIE 0.3.0","text":" - Lookup table for hg38 to hg19 circRNA annotations is updated... this eliminate one-to-many hits from the previous version
- BSJs extracted as different bam file.
- flowchart added
- adding slurmjobid to log/err file names
- v0.3.1 has significant (>10X) performance improvements at BSJ bam creation
- v0.3.3 splits BSJ bams into human and viral bams, and also converts them to bigwigs
- v0.3.4 adds hg38_rRNA_masked_plus_rRNA_plus_viruses_plus_ERCC reference (source:Sarah)
"},{"location":"CHANGELOG/#charlie-020","title":"CHARLIE 0.2.0","text":" - SE support added .. PE/SE samples handled concurrently
envmodules used in Snakemake in place of module load statements
"},{"location":"CHANGELOG/#charlie-010","title":"CHARLIE 0.1.0","text":" - base version
- PE only support
"},{"location":"contributing/","title":"Contributing to CHARLIE","text":""},{"location":"contributing/#proposing-changes-with-issues","title":"Proposing changes with issues","text":"If you want to make a change, it's a good idea to first open an issue and make sure someone from the team agrees that it\u2019s needed.
If you've decided to work on an issue, assign yourself to the issue so others will know you're working on it.
"},{"location":"contributing/#pull-request-process","title":"Pull request process","text":"We use GitHub Flow as our collaboration process. Follow the steps below for detailed instructions on contributing changes to CHARLIE.
"},{"location":"contributing/#clone-the-repo","title":"Clone the repo","text":"If you are a member of CCBR, you can clone this repository to your computer or development environment. Otherwise, you will first need to fork the repo and clone your fork. You only need to do this step once.
git clone https://github.com/CCBR/CHARLIE\n
Cloning into 'CHARLIE'... remote: Enumerating objects: 1136, done. remote: Counting objects: 100% (463/463), done. remote: Compressing objects: 100% (357/357), done. remote: Total 1136 (delta 149), reused 332 (delta 103), pack-reused 673 Receiving objects: 100% (1136/1136), 11.01 MiB | 9.76 MiB/s, done. Resolving deltas: 100% (530/530), done.
cd CHARLIE\n
"},{"location":"contributing/#if-this-is-your-first-time-cloning-the-repo-you-may-need-to-install-dependencies","title":"If this is your first time cloning the repo, you may need to install dependencies","text":" -
Install snakemake and singularity or docker if needed (biowulf already has these available as modules).
-
Install the python dependencies with pip
pip install .\n
If you're developing on biowulf, you can use our shared conda environment which already has these dependencies installed
. \"/data/CCBR_Pipeliner/db/PipeDB/Conda/etc/profile.d/conda.sh\"\nconda activate py311\n
- Install
pre-commit if you don't already have it. Then from the repo's root directory, run
pre-commit install\n
This will install the repo's pre-commit hooks. You'll only need to do this step the first time you clone the repo.
"},{"location":"contributing/#create-a-branch","title":"Create a branch","text":"Create a Git branch for your pull request (PR). Give the branch a descriptive name for the changes you will make, such as iss-10 if it is for a specific issue.
# create a new branch and switch to it\ngit branch iss-10\ngit switch iss-10\n
Switched to a new branch 'iss-10'
"},{"location":"contributing/#make-your-changes","title":"Make your changes","text":"Edit the code, write and run tests, and update the documentation as needed.
"},{"location":"contributing/#test","title":"test","text":"Changes to the python package code will also need unit tests to demonstrate that the changes work as intended. We write unit tests with pytest and store them in the tests/ subdirectory. Run the tests with python -m pytest.
If you change the workflow, please run the workflow with the test profile and make sure your new feature or bug fix works as intended.
"},{"location":"contributing/#document","title":"document","text":"If you have added a new feature or changed the API of an existing feature, you will likely need to update the documentation in docs/.
"},{"location":"contributing/#commit-and-push-your-changes","title":"Commit and push your changes","text":"If you're not sure how often you should commit or what your commits should consist of, we recommend following the \"atomic commits\" principle where each commit contains one new feature, fix, or task. Learn more about atomic commits here: https://www.freshconsulting.com/insights/blog/atomic-commits/
First, add the files that you changed to the staging area:
git add path/to/changed/files/\n
Then make the commit. Your commit message should follow the Conventional Commits specification. Briefly, each commit should start with one of the approved types such as feat, fix, docs, etc. followed by a description of the commit. Take a look at the Conventional Commits specification for more detailed information about how to write commit messages.
git commit -m 'feat: create function for awesome feature'\n
pre-commit will enforce that your commit message and the code changes are styled correctly and will attempt to make corrections if needed.
Check for added large files..............................................Passed Fix End of Files.........................................................Passed Trim Trailing Whitespace.................................................Failed
- hook id: trailing-whitespace
- exit code: 1
- files were modified by this hook > Fixing path/to/changed/files/file.txt > codespell................................................................Passed style-files..........................................(no files to check)Skipped readme-rmd-rendered..................................(no files to check)Skipped use-tidy-description.................................(no files to check)Skipped
In the example above, one of the hooks modified a file in the proposed commit, so the pre-commit check failed. You can run git diff to see the changes that pre-commit made and git status to see which files were modified. To proceed with the commit, re-add the modified file(s) and re-run the commit command:
git add path/to/changed/files/file.txt\ngit commit -m 'feat: create function for awesome feature'\n
This time, all the hooks either passed or were skipped (e.g. hooks that only run on R code will not run if no R files were committed). When the pre-commit check is successful, the usual commit success message will appear after the pre-commit messages showing that the commit was created.
Check for added large files..............................................Passed Fix End of Files.........................................................Passed Trim Trailing Whitespace.................................................Passed codespell................................................................Passed style-files..........................................(no files to check)Skipped readme-rmd-rendered..................................(no files to check)Skipped use-tidy-description.................................(no files to check)Skipped Conventional Commit......................................................Passed > [iss-10 9ff256e] feat: create function for awesome feature 1 file changed, 22 insertions(+), 3 deletions(-)
Finally, push your changes to GitHub:
git push\n
If this is the first time you are pushing this branch, you may have to explicitly set the upstream branch:
git push --set-upstream origin iss-10\n
Enumerating objects: 7, done. Counting objects: 100% (7/7), done. Delta compression using up to 10 threads Compressing objects: 100% (4/4), done. Writing objects: 100% (4/4), 648 bytes | 648.00 KiB/s, done. Total 4 (delta 3), reused 0 (delta 0), pack-reused 0 remote: Resolving deltas: 100% (3/3), completed with 3 local objects. remote: remote: Create a pull request for 'iss-10' on GitHub by visiting: remote: https://github.com/CCBR/CHARLIE/pull/new/iss-10 remote: To https://github.com/CCBR/CHARLIE > > [new branch] iss-10 -> iss-10 branch 'iss-10' set up to track 'origin/iss-10'.
We recommend pushing your commits often so they will be backed up on GitHub. You can view the files in your branch on GitHub at https://github.com/CCBR/CHARLIE/tree/<your-branch-name> (replace <your-branch-name> with the actual name of your branch).
"},{"location":"contributing/#create-the-pr","title":"Create the PR","text":"Once your branch is ready, create a PR on GitHub: https://github.com/CCBR/CHARLIE/pull/new/
Select the branch you just pushed:
Edit the PR title and description. The title should briefly describe the change. Follow the comments in the template to fill out the body of the PR, and you can delete the comments (everything between <!-- and -->) as you go. Be sure to fill out the checklist, checking off items as you complete them or striking through any irrelevant items. When you're ready, click 'Create pull request' to open it.
Optionally, you can mark the PR as a draft if you're not yet ready for it to be reviewed, then change it later when you're ready.
"},{"location":"contributing/#wait-for-a-maintainer-to-review-your-pr","title":"Wait for a maintainer to review your PR","text":"We will do our best to follow the tidyverse code review principles: https://code-review.tidyverse.org/. The reviewer may suggest that you make changes before accepting your PR in order to improve the code quality or style. If that's the case, continue to make changes in your branch and push them to GitHub, and they will appear in the PR.
Once the PR is approved, the maintainer will merge it and the issue(s) the PR links will close automatically. Congratulations and thank you for your contribution!
"},{"location":"contributing/#after-your-pr-has-been-merged","title":"After your PR has been merged","text":"After your PR has been merged, update your local clone of the repo by switching to the main branch and pulling the latest changes:
git checkout main\ngit pull\n
It's a good idea to run git pull before creating a new branch so it will start from the most recent commits in main.
"},{"location":"contributing/#helpful-links-for-more-information","title":"Helpful links for more information","text":" - GitHub Flow
- semantic versioning guidelines
- changelog guidelines
- tidyverse code review principles
- reproducible examples
- nf-core extensions for VS Code
"},{"location":"flowchart/","title":"CHARLIE workflow","text":"DISCLAIMER: This chart is for v0.8.x may be slightly outdated.
"},{"location":"platforms/","title":"Platforms","text":"CHARLIE was originally developed to run on biowulf, but it can run on other computing platforms too. There are a few additional steps to configure CHARLIE.
TODO
- Clone CHARLIE.
git clone https://github.com/CCBR/charlie\n
- Initialize your project working directory.
\n
-
Create a directory of reference files.
-
Edit your project's config file.
-
If you are using a SLURM job scheduler, edit cluster.json and submit_script.sbatch.
"},{"location":"references/","title":"References","text":""},{"location":"references/#references","title":"References","text":"The reference sequences comprises of the host genome and the viral genomes.
"},{"location":"references/#fasta","title":"Fasta","text":"hg38 and mm39 genome builds are chosen to represent hosts. Ribosomal sequences (45S, 5S) are downloaded from NCBI. hg38 and mm39 were masked for rRNA sequence and 45S and 5S sequences from NCBI are appended as separate chromosomes. The following viral sequences were appended to the rRNA masked hg38 reference:
HOSTS:\n * hg38 [Human]\n * mm39 [Mouse]\n\nADDITIVES:\n * ERCC [External RNA Control Consortium sequences]\n * BAC16Insert [insert from rKSHV.219-derived BAC clone of the full-length KSHV genome]\n\nVIRUSES:\n * NC_007605.1 [Human gammaherpesvirus 4 (Epstein-Barr virus)]\n * NC_006273.2 [Human betaherpesvirus 5 (Cytomegalovirus )]\n * NC_001664.4 [Human betaherpesvirus 6A (HHV-6A)]\n * NC_000898.1 [Human betaherpesvirus 6B (HHV-6B)]\n * NC_001716.2 [Human betaherpesvirus 7 (HHV-7)]\n * NC_009333.1 [Human gammaherpesvirus 8 (KSHV)]\n * NC_045512.2 [Severe acute respiratory syndrome(SARS)-related coronavirus]\n * MN485971.1 [HIV from Belgium]\n * NC_001806.2 [Human alphaherpesvirus 1 (Herpes simplex virus type 1)](strain 17) (HSV-1)]\n * KT899744.1 [HSV-1 strain KOS]\n * MH636806.1 [MHV68 (Murine herpesvirus 68 strain WUMS)]\n
Location: The entire resource bundle is available at /data/CCBR_Pipeliner/db/PipeDB/charlie/fastas_gtfs on BIOWULF. This location also have additional bash scripts required for aggregating annotations and building indices required by different aligners.
When -m=dryrun is run for the first time after initialization (-m=init), the appropriate host+additives+viruses fasta and gtf files are created on the fly, which are then used to build aligner reference indexes automatically.
"},{"location":"tutorial/","title":"Tutorial","text":""},{"location":"tutorial/#prerequisites","title":"Prerequisites","text":" - Biowulf account: Biowulf account can be requested here.
"},{"location":"tutorial/#installation","title":"Installation","text":"CHARLIE is already installed on biowulf. It is included in the ccbrpipeliner module from release 7 onward. To load the module run:
module load ccbrpipeliner/7\n
"},{"location":"tutorial/#init","title":"Init","text":"To get help about the pipeline you can run:
charlie --help\n
output:
##########################################################################################\n\nWelcome to charlie(v0.10.0-dev)\n _______ __ __ _______ ______ ___ ___ _______\n| || | | || _ || _ | | | | | | |\n| || |_| || |_| || | || | | | | | ___|\n| || || || |_||_ | | | | | |___\n| _|| || || __ || |___ | | | ___|\n| |_ | _ || _ || | | || || | | |___\n|_______||__| |__||__| |__||___| |_||_______||___| |_______|\n\nC_ircrnas in H_ost A_nd vi_R_uses ana_L_ysis p_I_p_E_line\n\n##########################################################################################\n\nThis pipeline was built by CCBR (https://bioinformatics.ccr.cancer.gov/ccbr)\nPlease contact Vishal Koparde for comments/questions (vishal.koparde@nih.gov)\n\n##########################################################################################\n\nCHARLIE can be used to DAQ (Detect/Annotate/Quantify) circRNAs in hosts and viruses.\n\nHere is the list of hosts and viruses that are currently supported:\n\nHOSTS:\n * hg38 [Human]\n * mm39 [Mouse]\n\nADDITIVES:\n * ERCC [External RNA Control Consortium sequences]\n * BAC16Insert [insert from rKSHV.219-derived BAC clone of the full-length KSHV genome]\n\nVIRUSES:\n * NC_007605.1 [Human gammaherpesvirus 4 (Epstein-Barr virus)]\n * NC_006273.2 [Human betaherpesvirus 5 (Cytomegalovirus )]\n * NC_001664.4 [Human betaherpesvirus 6A (HHV-6A)]\n * NC_000898.1 [Human betaherpesvirus 6B (HHV-6B)]\n * NC_001716.2 [Human betaherpesvirus 7 (HHV-7)]\n * NC_009333.1 [Human gammaherpesvirus 8 (KSHV)]\n * NC_045512.2 [Severe acute respiratory syndrome(SARS)-related coronavirus]\n * MN485971.1 [HIV from Belgium]\n * NC_001806.2 [Human alphaherpesvirus 1 (Herpes simplex virus type 1)](strain 17) (HSV-1)]\n * KT899744.1 [HSV-1 strain KOS]\n * MH636806.1 [MHV68 (Murine herpesvirus 68 strain WUMS)]\n\n##########################################################################################\n\nUSAGE:\n charlie -w/--workdir=<WORKDIR> -m/--runmode=<RUNMODE>\n\nRequired Arguments:\n1. WORKDIR : [Type: String]: Absolute or relative path to the output folder with write permissions.\n\n2. RUNMODE : [Type: String] Valid options:\n * init : initialize workdir\n * dryrun : dry run snakemake to generate DAG\n * run : run with slurm\n * runlocal : run without submitting to sbatch\n ADVANCED RUNMODES (use with caution!!)\n * unlock : unlock WORKDIR if locked by snakemake NEVER UNLOCK WORKDIR WHERE PIPELINE IS CURRENTLY RUNNING!\n * reconfig : recreate config file in WORKDIR (debugging option) EDITS TO config.yaml WILL BE LOST!\n * reset : DELETE workdir dir and re-init it (debugging option) EDITS TO ALL FILES IN WORKDIR WILL BE LOST!\n * printbinds: print singularity binds (paths)\n * local : same as runlocal\n\nOptional Arguments:\n\n--singcache|-c : singularity cache directory. Default is `/data/${USER}/.singularity` if available, or falls back to `${WORKDIR}/.singularity`. Use this flag to specify a different singularity cache directory.\n--host|-g : supply host at command line. hg38 or mm39. (--runmode=init only)\n--additives|-a : supply comma-separated list of additives at command line. ERCC or BAC16Insert or both (--runmode=init only)\n--viruses|-v : supply comma-separated list of viruses at command line (--runmode=init only)\n--manifest|-s : absolute path to samples.tsv. This will be copied to output folder (--runmode=init only)\n--changegrp|-z : change group to \"Ziegelbauer_lab\" before running anything. Biowulf-only. Useful for correctly setting permissions.\n--help|-h : print this help\n\n\nExample commands:\n charlie -w=/my/output/folder -m=init\n charlie -w=/my/output/folder -m=dryrun\n charlie -w=/my/output/folder -m=run\n\n##########################################################################################\n\nVersionInfo:\n python : 3\n snakemake : 7\n pipeline_home : /gpfs/gsfs10/users/CCBR_Pipeliner/Pipelines/CHARLIE/.v0.11.1\n git commit/tag : 613fb617f1ed426fb8900f98e599ca0497a67cc0 v0.11.0-49-g613fb61\n\n##########################################################################################\n
NOTE: You can replace v0.10.0 in the above command with the latest version to use a newer version. run_circrna_daq.sh was called test.sh in versions older than v0.4.0.
To initial the working directory run:
charlie -w=<path to output dir> -m=init\n
This assumes that <path to output dir> does not exist before running the above command and is at a location where write permissions are available.
The above command creates <path to output dir> folder and creates 2 subfolders logs and stats inside that folder along with config.yaml and samples.tsv files.
tree <path to output dir>\n
"},{"location":"tutorial/#configyaml","title":"config.yaml","text":"This file is used to fine tune the execution of the pipeline by setting:
- sample sheet location ... aka
samples.tsv - the temporary directory -- make sure this is correct for your computing environment.
- which circRNA finding tools to use by editing these:
- run_clear: True
- run_dcc: True
- run_mapsplice: False
- run_circRNAFinder: True
- run_nclscan: False
- run_findcirc: False
- describes the location of other resources/indexes/tools etc. Generally, these do NOT need to be changed.
"},{"location":"tutorial/#samplestsv","title":"samples.tsv","text":"Tab delimited definition of sample sheet. The header is fixed and each row represents a sample. It has 3 columns:
- sampleName = Name of the sample. This has to be unique.
- path_to_R1_fastq = absolute path to the read1 fastq.gz file.
- path_to_R2_fastq = absolute path to the read2 fastq.gz file. If the sample was sequenced in single-end mode, then leave this blank.
The /data/CCBR_Pipeliner/testdata/circRNA/humans folder in the repo has test dataset:
tree /data/CCBR_Pipeliner/testdata/circRNA/humans\n/data/CCBR_Pipeliner/testdata/circRNA/humans\n\u251c\u2500\u2500 GI1_N_ss.R1.fastq.gz\n\u251c\u2500\u2500 GI1_N_ss.R2.fastq.gz\n\u251c\u2500\u2500 GI1_T_ss.R1.fastq.gz\n\u2514\u2500\u2500 samples.tsv\n
GI1_N is a PE sample while GI1_T is a SE sample.
"},{"location":"tutorial/#dryrun","title":"Dryrun","text":"Once the samples.tsv file has been edited appropriately to include the desired samples, it is a good idea to dryrun the pipeline to ensure that everything will work as desired. Dryrun can be run as follows:
charlie -w=<path to output dir> -m=dryrun\n
This will create the reference fasta and gtf file based on the selections made in the config.yaml. Hence, can take a few minutes to run.
"},{"location":"tutorial/#run","title":"Run","text":"Upon verifying that dryrun is successful. You can then submit the job to the cluster using the following command:
charlie -w=<path to output dir> -m=run\n
which will produce something like this:
... ... skipping ~1000 lines\n...\n...\nJob stats:\njob count min threads max threads\n--------------------------------------------- ------- ----------\nall 1 1 1\nannotate_clear_output 2 1 1\ncircExplorer 2 2 2\ncircExplorer_bwa 2 2 2\ncircrnafinder 2 1 1\nciri 2 56 56\nclear 2 2 2\ncreate_bowtie2_index 1 1 1\ncreate_bwa_index 1 1 1\ncreate_circExplorer_BSJ_bam 2 4 4\ncreate_circExplorer_linear_spliced_bams 2 56 56\ncreate_circExplorer_merged_found_counts_table 2 1 1\ncreate_hq_bams 2 1 1\ncreate_index 1 56 56\ncreate_master_counts_file 1 1 1\ncutadapt 2 56 56\ndcc 2 4 4\ndcc_create_samplesheets 2 1 1\nestimate_duplication 2 1 1\nfastqc 2 4 4\nfind_circ 2 56 56\nfind_circ_align 2 56 56\nmerge_SJ_tabs 1 2 2\nmerge_alignment_stats 1 1 1\nmerge_genecounts 1 1 1\nmerge_per_sample 2 1 1\nstar1p 2 56 56\nstar2p 2 56 56\nstar_circrnafinder 2 56 56\ntotal 52 1 56\n\nReasons:\n (check individual jobs above for details)\n input files updated by another job:\n alignment_stats, all, annotate_clear_output, circExplorer, circExplorer_bwa, circrnafinder, ciri, clear, create_circExplorer_BSJ_bam, create_circExplorer_linear_spliced_bams, create_circExplorer_merged_found_counts_table, create_hq_bams, create_master_counts_file, dcc, dcc_create_samplesheets, estimate_duplication, fastqc, find_circ, find_circ_align, merge_SJ_tabs, merge_alignment_stats, merge_genecounts, merge_per_sample, star1p, star2p, star_circrnafinder\n missing output files:\n alignment_stats, annotate_clear_output, circExplorer, circExplorer_bwa, circrnafinder, ciri, clear, create_bowtie2_index, create_bwa_index, create_circExplorer_BSJ_bam, create_circExplorer_linear_spliced_bams, create_circExplorer_merged_found_counts_table, create_hq_bams, create_index, create_master_counts_file, cutadapt, dcc, dcc_create_samplesheets, estimate_duplication, fastqc, find_circ, find_circ_align, merge_SJ_tabs, merge_alignment_stats, merge_genecounts, merge_per_sample, star1p, star2p, star_circrnafinder\n\nThis was a dry-run (flag -n). The order of jobs does not reflect the order of execution.\nRunning...\n14743440\n
In this example, 14743440 is the jobid returned by the slurm job scheduler on biowulf. This means that the job was successfully submitted, it will spawn off other subjobs which in-turn will be run and outputs will be moved to the results folder created inside the working directory supplied at command line. You can check the status of your queue of jobs in biowulf running:
squeue -u `whoami`\n
output:
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)\n 14743440 ccr,norm circRNA kopardev PD 0:00 1 (None)\n
ST in the above results stands for Status and PD means Pending. The status will change from pending(PD) to running(R) to completed as jobs are run on the cluster.
Next, just sit tight until the pipeline finishes. You can keep monitoring the queue as shown above. If there are no jobs running on biowulf, then your pipeline has finished (or errored out!)
Once completed the output should something like this:
tree <path to output dir>\n
output:
.\n\u251c\u2500\u2500 cluster.json\n\u251c\u2500\u2500 config.yaml\n\u251c\u2500\u2500 dryrun.231222103505.log\n\u251c\u2500\u2500 fastqs\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R1.fastq.gz -> /data/CCBR_Pipeliner/testdata/circRNA/human/GI1_N_ss.R1.fastq.gz\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R2.fastq.gz -> /data/CCBR_Pipeliner/testdata/circRNA/human/GI1_N_ss.R2.fastq.gz\n\u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T.R1.fastq.gz -> /data/CCBR_Pipeliner/testdata/circRNA/human/GI1_T_ss.R1.fastq.gz\n\u251c\u2500\u2500 logs/snakemake.log.jobby.short\n\u251c\u2500\u2500 logs/snakemake.log.jobby.txt\n\u251c\u2500\u2500 logs\n\u2502 ... log files ...\n\u2502 ... skipping ...\n\u251c\u2500\u2500 nclscan.config\n\u251c\u2500\u2500 qc\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 fastqc\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R1_fastqc.html\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R1_fastqc.zip\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R1.trim_fastqc.html\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R1.trim_fastqc.zip\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R2_fastqc.html\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R2_fastqc.zip\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R2.trim_fastqc.html\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R2.trim_fastqc.zip\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.R1_fastqc.html\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.R1_fastqc.zip\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.R1.trim_fastqc.html\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T.R1.trim_fastqc.zip\n\u2502\u00a0\u00a0 \u2514\u2500\u2500 picard_MarkDuplicates\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.MarkDuplicates.metrics.txt\n\u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T.MarkDuplicates.metrics.txt\n\u251c\u2500\u2500 ref\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 bwa_index.log\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 gene_id_2_gene_name.tsv\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 NCLscan_index\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 AllRef.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 AllRef.fa.amb\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 AllRef.fa.ann\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 AllRef.fa.bwt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 AllRef.fa.pac\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 AllRef.fa.sa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 AllRef.ndx\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 RepChrM.fa\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.1.bt2\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.2.bt2\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.3.bt2\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.4.bt2\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.amb\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.ann\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.bwt\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.dummy.fa\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.fa\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.fa.byo_index\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.fa.fai\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.fa.regions\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.fa.regions.host\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.fa.regions.viruses\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.fa.sizes\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.fixed.gtf\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.genes.genepred\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.genes.genepred_w_geneid\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.gtf\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.pac\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.rev.1.bt2\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.rev.2.bt2\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.sa\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 ref.transcripts.fa\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 separate_fastas\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr10.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr11.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr12.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr13.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr14.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr15.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr16.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr17.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr18.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr19.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr20.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr21.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr22.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr2.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr3.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr45S.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr4.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr5.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr5S.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr6.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr7.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr8.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chr9.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chrM.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chrX.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 chrY.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00002.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00003.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00004.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00009.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00012.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00013.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00014.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00016.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00017.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00019.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00022.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00024.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00025.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00028.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00031.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00033.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00034.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00035.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00039.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00040.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00041.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00042.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00043.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00044.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00046.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00048.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00051.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00053.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00054.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00057.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00058.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00059.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00060.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00061.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00062.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00067.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00069.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00071.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00073.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00074.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00075.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00076.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00077.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00078.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00079.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00081.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00083.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00084.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00085.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00086.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00092.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00095.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00096.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00097.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00098.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00099.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00104.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00108.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00109.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00111.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00112.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00113.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00116.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00117.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00120.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00123.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00126.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00130.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00131.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00134.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00136.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00137.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00138.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00142.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00143.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00144.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00145.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00147.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00148.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00150.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00154.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00156.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00157.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00158.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00160.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00162.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00163.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00164.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00165.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00168.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00170.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ERCC_00171.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000008.2.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000009.2.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000194.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000195.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000205.2.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000208.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000213.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000214.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000216.2.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000218.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000219.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000220.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000221.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000224.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000225.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GL000226.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270302.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270303.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270304.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270305.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270310.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270311.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270312.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270315.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270316.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270317.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270320.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270322.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270329.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270330.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270333.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270334.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270335.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270336.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270337.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270338.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270340.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270362.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270363.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270364.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270366.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270371.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270372.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270373.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270374.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270375.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270376.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270378.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270379.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270381.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270382.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270383.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270384.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270385.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270386.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270387.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270388.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270389.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270390.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270391.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270392.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270393.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270394.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270395.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270396.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270411.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270412.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270414.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270417.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270418.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270419.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270420.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270422.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270423.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270424.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270425.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270429.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270435.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270438.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270442.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270448.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270465.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270466.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270467.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270468.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270507.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270508.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270509.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270510.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270511.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270512.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270515.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270516.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270517.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270518.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270519.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270521.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270522.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270528.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270529.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270530.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270538.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270539.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270544.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270548.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270579.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270580.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270581.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270582.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270583.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270584.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270587.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270588.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270589.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270590.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270591.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270593.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270706.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270707.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270708.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270709.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270710.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270711.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270712.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270713.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270714.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270715.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270716.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270717.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270718.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270719.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270720.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270721.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270722.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270723.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270724.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270725.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270726.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270727.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270728.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270729.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270730.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270731.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270732.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270733.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270734.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270735.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270736.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270737.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270738.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270739.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270740.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270741.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270742.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270743.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270744.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270745.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270746.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270747.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270748.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270749.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270750.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270751.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270752.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270753.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270754.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270755.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270756.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 KI270757.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 NC_009333.1.fa\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 separate_fastas.lst\n\u2502\u00a0\u00a0 \u2514\u2500\u2500 STAR_no_GTF\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 chrLength.txt\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 chrNameLength.txt\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 chrName.txt\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 chrStart.txt\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 Genome\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 genomeParameters.txt\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 Log.out\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 SA\n\u2502\u00a0\u00a0 \u2514\u2500\u2500 SAindex\n\u251c\u2500\u2500 results\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 alignmentstats.txt\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 circRNA_master_counts.tsv.gz\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 alignmentstats.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 circExplorer\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.back_spliced_junction.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.back_spliced_junction.strand_fixed.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.bam.bai\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.bed.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.foundcounts.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.minus.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.minus.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.minus.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.plus.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.plus.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.BSJ.plus.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.circExplorer.annotation_counts.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.circExplorer.counts_table.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_circexplorer_parse.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.circRNA_known.filter1.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.circularRNA_known.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.hg38.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.hg38.BSJ.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.hg38.BSJ.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear_BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear_BSJ.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear_BSJ.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear_BSJ.hg38.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear_BSJ.hg38.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear_BSJ.NC_009333.1.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear_BSJ.NC_009333.1.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear.BSJ.readids.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear.hg38.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear.hg38.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear.NC_009333.1.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear.NC_009333.1.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.linear_spliced.counts.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.NC_009333.1.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.NC_009333.1.BSJ.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.NC_009333.1.BSJ.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.readcounts.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.rid2jid.tsv.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced_BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced_BSJ.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced_BSJ.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced_BSJ.hg38.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced_BSJ.hg38.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced_BSJ.NC_009333.1.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced_BSJ.NC_009333.1.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced.BSJ.readids.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced.hg38.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced.hg38.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced.NC_009333.1.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.spliced.NC_009333.1.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 low_conf_GI1_N.circRNA_known.filter1.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 low_conf_GI1_N.circRNA_known.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 circExplorer_BWA\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.back_spliced_junction.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.back_spliced_junction.strand_fixed.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.circExplorer_bwa.annotation_counts.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_circexplorer_parse.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.circRNA_known.filter1.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.circularRNA_known.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 low_conf_GI1_N.circRNA_known.filter1.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 low_conf_GI1_N.circRNA_known.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 circRNA_finder\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.Chimeric.out.sorted.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.Chimeric.out.sorted.bam.bai\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.circRNA_finder.counts_table.tsv.filtered\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.filteredJunctions.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.s_filteredJunctions.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_N.s_filteredJunctions_fw.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ciri\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 CIRIerror.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.bwa.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.ciri.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.ciri.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.ciri.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.ciri.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_N.ciri.out.filtered\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 CLEAR\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 quant.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 quant.txt.annotated\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 DCC\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 CircCoordinates\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 CircRNACount\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 CircSkipJunctions\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 DCC-2023-12-20_1149.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.dcc.counts_table.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.dcc.counts_table.tsv.filtered\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 LinearCount\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 mate1.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 mate2.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 samplesheet.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 find_circ\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_anchors.fastq.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.unmapped.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_N.unmapped.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.circRNA_counts.txt.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 merge_per_sample.sh\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 STAR1p\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p1.Chimeric.out.junction\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p1.Log.final.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p1.Log.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p1.Log.progress.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p1.SJ.out.tab\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 mate1\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_mate1.Chimeric.out.junction\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_mate1.Log.final.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_mate1.Log.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_mate1.Log.progress.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_N_mate1.SJ.out.tab\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 mate2\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_mate2.Chimeric.out.junction\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_mate2.Chimeric.out.junction.fixed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_mate2.Log.final.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_mate2.Log.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_mate2.Log.progress.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_N_mate2.SJ.out.tab\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 STAR2p\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.chimeric.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.chimeric.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.Chimeric.out.junction\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.Log.final.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.Log.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.Log.progress.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.non_chimeric.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.non_chimeric.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N_p2.ReadsPerGene.out.tab\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_N_p2.SJ.out.tab\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 STAR_circRNAFinder\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.Chimeric.out.junction\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.Chimeric.out.sam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.Log.final.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.Log.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.Log.progress.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_N.SJ.out.tab\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 trim\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.R1.trim.fastq.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_N.R2.trim.fastq.gz\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 alignmentstats.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 circExplorer\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.back_spliced_junction.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.back_spliced_junction.strand_fixed.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.bam.bai\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.bed.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.foundcounts.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.minus.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.minus.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.minus.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.plus.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.plus.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.BSJ.plus.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.circExplorer.annotation_counts.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.circExplorer.counts_table.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_circexplorer_parse.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.circRNA_known.filter1.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.circularRNA_known.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.hg38.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.hg38.BSJ.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.hg38.BSJ.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear_BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear_BSJ.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear_BSJ.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear_BSJ.hg38.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear_BSJ.hg38.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear_BSJ.NC_009333.1.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear_BSJ.NC_009333.1.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear.BSJ.readids.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear.hg38.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear.hg38.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear.NC_009333.1.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear.NC_009333.1.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.linear_spliced.counts.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.NC_009333.1.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.NC_009333.1.BSJ.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.NC_009333.1.BSJ.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.readcounts.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.rid2jid.tsv.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced_BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced_BSJ.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced_BSJ.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced_BSJ.hg38.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced_BSJ.hg38.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced_BSJ.NC_009333.1.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced_BSJ.NC_009333.1.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced.BSJ.readids.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced.hg38.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced.hg38.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced.NC_009333.1.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.spliced.NC_009333.1.bw\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 low_conf_GI1_T.circRNA_known.filter1.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 low_conf_GI1_T.circRNA_known.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 circExplorer_BWA\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.back_spliced_junction.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.back_spliced_junction.strand_fixed.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.circExplorer_bwa.annotation_counts.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_circexplorer_parse.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.circRNA_known.filter1.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.circularRNA_known.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 low_conf_GI1_T.circRNA_known.filter1.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 low_conf_GI1_T.circRNA_known.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 circRNA_finder\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.Chimeric.out.sorted.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.Chimeric.out.sorted.bam.bai\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.circRNA_finder.counts_table.tsv.filtered\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.filteredJunctions.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.s_filteredJunctions.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T.s_filteredJunctions_fw.bed\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 ciri\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 CIRIerror.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.bwa.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.ciri.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.ciri.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.ciri.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.ciri.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T.ciri.out.filtered\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 CLEAR\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 quant.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 quant.txt.annotated\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 DCC\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 CircCoordinates\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 CircRNACount\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 CircSkipJunctions\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 DCC-2023-12-20_1206.log\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.dcc.counts_table.tsv\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.dcc.counts_table.tsv.filtered\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 LinearCount\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 mate1.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 mate2.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 samplesheet.txt\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 find_circ\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_anchors.fastq.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.unmapped.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T.unmapped.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.circRNA_counts.txt.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 merge_per_sample.sh\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 STAR1p\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p1.Chimeric.out.junction\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p1.Chimeric.out.junction.circRNA\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p1.Chimeric.out.junction.circRNAmapped\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p1.Log.final.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p1.Log.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p1.Log.progress.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p1.SJ.out.tab\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 mate1\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T_mate1.Chimeric.out.junction\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 mate2\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T_mate2.Chimeric.out.junction\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 STAR2p\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.chimeric.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.chimeric.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.Chimeric.out.junction\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.Log.final.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.Log.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.Log.progress.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.non_chimeric.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.non_chimeric.bam.csi\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T_p2.ReadsPerGene.out.tab\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T_p2.SJ.out.tab\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 STAR_circRNAFinder\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.Chimeric.out.junction\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.Chimeric.out.sam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.Log.final.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.Log.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.Log.progress.out\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T.SJ.out.tab\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 trim\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.R1.trim.fastq.gz\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T.R2.trim.fastq.gz\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 HQ_BSJ_bams\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.hg38.HQ_only.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.hg38.HQ_only.BSJ.bam.bai\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.HQ_only.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.HQ_only.BSJ.bam.bai\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.NC_009333.1.HQ_only.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_N.NC_009333.1.HQ_only.BSJ.bam.bai\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.hg38.HQ_only.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.hg38.HQ_only.BSJ.bam.bai\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.HQ_only.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.HQ_only.BSJ.bam.bai\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u251c\u2500\u2500 GI1_T.NC_009333.1.HQ_only.BSJ.bam\n\u2502\u00a0\u00a0 \u2502\u00a0\u00a0 \u2514\u2500\u2500 GI1_T.NC_009333.1.HQ_only.BSJ.bam.bai\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 pass1.out.tab\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 revstranded_STAR_GeneCounts.tsv\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 stranded_STAR_GeneCounts.tsv\n\u2502\u00a0\u00a0 \u2514\u2500\u2500 unstranded_STAR_GeneCounts.tsv\n\u251c\u2500\u2500 runinfo.yaml\n\u251c\u2500\u2500 runslurm_snakemake_report.html\n\u251c\u2500\u2500 samples.tsv\n\u251c\u2500\u2500 snakemake.log\n\u251c\u2500\u2500 snakemake.stats\n\u251c\u2500\u2500 stats\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 dryrun.231222102114.log\n\u2502\u00a0\u00a0 \u251c\u2500\u2500 runinfo.modtime.yaml\n\u2502\u00a0\u00a0 \u2514\u2500\u2500 snakemake.20231222104214.log\n\u251c\u2500\u2500 submit_script.sbatch\n\u2514\u2500\u2500 tools.txt\n
"},{"location":"tutorial/#error-tracking","title":"Error tracking","text":"Using the following command to find FAILED jobs:
grep FAIL logs/snakemake.log.jobby.short\n
The above command also gives .err and .out log files which can give further insights on reasons for failure and changes required to be made for a successful run.
"},{"location":"tutorial/#expected-output","title":"Expected output:","text":"The main output file is results/circRNA_master_counts.tsv.gz. Here are the top 3 tiles from an example output:
Column_number Column_title Example_1 Example_2 Example_3 1 chrom GL000220.1 GL000220.1 GL000220.1 2 start 107635 112482 118578 3 end 151634 156427 118759 4 circExplorer_strand -1 -1 -1 5 circExplorer_bwa_strand . . . 6 ciri_strand -1 -1 -1 7 dcc_strand -1 -1 -1 8 circrnafinder_strand -1 -1 -1 9 flankingsites+ CC##GC GC##CC CC##GC 10 flankingsites- GG##GC GC##GG GG##GC 11 sample_name GI1_N GI1_N GI1_N 12 ntools 1 1 1 13 HQ N N N 14 circExplorer_read_count -1 -1 -1 15 circExplorer_found_BSJcounts -1 -1 -1 16 circExplorerfound_linear_BSJ+_counts -1 -1 -1 17 circExplorerfound_linear_spliced_BSJ+_counts -1 -1 -1 18 circExplorerfound_linear_BSJ-_counts -1 -1 -1 19 circExplorerfound_linear_spliced_BSJ-_counts -1 -1 -1 20 circExplorerfound_linear_BSJ._counts -1 -1 -1 21 circExplorerfound_linear_spliced_BSJ._counts -1 -1 -1 22 ciri_read_count -1 -1 -1 23 ciri_linear_read_count -1 -1 -1 24 circExplorer_bwa_read_count 3 7 3 25 dcc_read_count -1 -1 -1 26 dcc_linear_read_count -1 -1 -1 27 circrnafinder_read_count -1 -1 -1 28 hqcounts 1 1 1 29 nonhqcounts 0 0 0 30 circExplorer_annotation Unknown Unknown Unknown 31 ciri_annotation Unknown Unknown Unknown 32 circExplorer_bwa_annotation novel novel novel 33 dcc_gene Unknown Unknown Unknown 34 dcc_junction_type Unknown Unknown Unknown 35 dcc_annotation Unknown Unknown Unknown Expected output from the sample data is stored under .tests/expected_output.
"}]}
\ No newline at end of file
diff --git a/dev/sitemap.xml.gz b/dev/sitemap.xml.gz
index bd75785e9f55eebcb834795801164a4fb2c317ee..d7c33b061138be7b31fba75dfffbf7efe75417c0 100644
GIT binary patch
delta 13
Ucmb=gXP58h;9!t;oXB1Q02dYmEC2ui
delta 13
Ucmb=gXP58h;9$6FJ(0Zv02-hJ-~a#s
diff --git a/dev/tutorial/index.html b/dev/tutorial/index.html
index 70ff704..f70ad72 100644
--- a/dev/tutorial/index.html
+++ b/dev/tutorial/index.html
@@ -1,18 +1,5 @@
- Tutorial - CHARLIE (Circrnas in Host And viRuses anaLysis pIpEline) Tutorial¶
Prerequisites¶
-
-
Membership to Ziegelbauer user group on Biowulf. You can check this by typing the following command:
% groups
-
output:
CCBR kopardevn Ziegelbauer_lab
-
If Ziegelbauer_lab is not listed then you can email a request to be added to the groups here
Location¶
Different versions of circRNA DAQ pipeline have been parked at /data/Ziegelbauer_lab/Pipelines/circRNA
% ls /data/Ziegelbauer_lab/Pipelines/circRNA
-
output:
v0.1.0
-v0.10.0
-v0.10.0-dev
-v0.2.1
-v0.3.3
-v0.4.2
-v0.5.2
-v0.6.5
-v0.7.0
-v0.8
-v0.9.0
-
The exacts versions listed here may changed as newer versions are added. Also, the dev version is pointing to the most recent untagged version of the pipeline (use at own risk!)
Init¶
To get help about the pipeline you can run:
% bash /data/Ziegelbauer_lab/Pipelines/circRNA/v0.10.0-dev/charlie
+ Tutorial - CHARLIE (Circrnas in Host And viRuses anaLysis pIpEline) Tutorial¶
Prerequisites¶
Installation¶
CHARLIE is already installed on biowulf. It is included in the ccbrpipeliner module from release 7 onward. To load the module run:
module load ccbrpipeliner/7
+
Init¶
To get help about the pipeline you can run:
charlie --help
output:
##########################################################################################
Welcome to charlie(v0.10.0-dev)
@@ -33,7 +20,7 @@
##########################################################################################
-CHARLIE can be used to DAQ(Detect/Annotate/Quantify) circRNAs in hosts and viruses.
+CHARLIE can be used to DAQ (Detect/Annotate/Quantify) circRNAs in hosts and viruses.
Here is the list of hosts and viruses that are currently supported:
@@ -61,7 +48,7 @@
##########################################################################################
USAGE:
- bash /data/Ziegelbauer_lab/Pipelines/circRNA/v0.10.0-dev/charlie -w/--workdir=<WORKDIR> -m/--runmode=<RUNMODE>
+ charlie -w/--workdir=<WORKDIR> -m/--runmode=<RUNMODE>
Required Arguments:
1. WORKDIR : [Type: String]: Absolute or relative path to the output folder with write permissions.
@@ -90,28 +77,29 @@
Example commands:
- bash /data/Ziegelbauer_lab/Pipelines/circRNA/v0.10.0-dev/charlie -w=/my/output/folder -m=init
- bash /data/Ziegelbauer_lab/Pipelines/circRNA/v0.10.0-dev/charlie -w=/my/output/folder -m=dryrun
- bash /data/Ziegelbauer_lab/Pipelines/circRNA/v0.10.0-dev/charlie -w=/my/output/folder -m=run
+ charlie -w=/my/output/folder -m=init
+ charlie -w=/my/output/folder -m=dryrun
+ charlie -w=/my/output/folder -m=run
##########################################################################################
VersionInfo:
- python : 3.7
- snakemake : 7.19.1
- pipeline_home : /vf/users/Ziegelbauer_lab/Pipelines/circRNA/v0.10.0-dev
- git commit/tag : b2cf2f089788651041b16bf4378c2c5172c13cb2 v0.10.0-2-gb2cf2f0
+ python : 3
+ snakemake : 7
+ pipeline_home : /gpfs/gsfs10/users/CCBR_Pipeliner/Pipelines/CHARLIE/.v0.11.1
+ git commit/tag : 613fb617f1ed426fb8900f98e599ca0497a67cc0 v0.11.0-49-g613fb61
##########################################################################################
-
NOTE: You can replace v0.10.0 in the above command with the latest version to use a newer version. run_circrna_daq.sh was called test.sh in versions older than v0.4.0.
To initial the working directory run:
% bash <path to charlie> -w=<path to output dir> -m=init
-
This assumes that <path to output dir> does not exist before running the above command and is at a location where write permissions are available.
The above command creates <path to output dir> folder and creates 2 subfolders logs and stats inside that folder along with config.yaml and samples.tsv files.
% tree <path to output dir>
-
config.yaml¶
This file is used to fine tune the execution of the pipeline by setting:
- sample sheet location ... aka
samples.tsv - the temporary directory -- make sure this is correct for your computing environment.
- which circRNA finding tools to use by editing these:
- run_clear: True
- run_dcc: True
- run_mapsplice: False
- run_circRNAFinder: True
- run_nclscan: False
- run_findcirc: False
- describes the location of other resources/indexes/tools etc. Generally, these do NOT need to be changed.
samples.tsv¶
Tab delimited definition of sample sheet. The header is fixed and each row represents a sample. It has 3 columns:
- sampleName = Name of the sample. This has to be unique.
- path_to_R1_fastq = absolute path to the read1 fastq.gz file.
- path_to_R2_fastq = absolute path to the read2 fastq.gz file. If the sample was sequenced in single-end mode, then leave this blank.
The .tests/dummy_fastqs folder in the repo has test dataset:
% tree .tests/dummy_fastqs
-.tests/dummy_fastqs
-├── GI1_N.R1.fastq.gz
-├── GI1_N.R2.fastq.gz
-└── GI1_T.R1.fastq.gz
-
GI1_N is a PE sample while GI1_T is a SE sample.
Dryrun¶
Once the samples.tsv file has been edited appropriately to include the desired samples, it is a good idea to dryrun the pipeline to ensure that everything will work as desired. Dryrun can be run as follows:
bash <path to charlie> -w=<path to output dir> -m=dryrun
-
This will create the reference fasta and gtf file based on the selections made in the config.yaml. Hence, can take a few minutes to run.
Run¶
Upon verifying that dryrun is successful. You can then submit the job to the cluster using the following command:
bash <path to charlie> -w=<path to output dir> -m=run
+
NOTE: You can replace v0.10.0 in the above command with the latest version to use a newer version. run_circrna_daq.sh was called test.sh in versions older than v0.4.0.
To initial the working directory run:
charlie -w=<path to output dir> -m=init
+
This assumes that <path to output dir> does not exist before running the above command and is at a location where write permissions are available.
The above command creates <path to output dir> folder and creates 2 subfolders logs and stats inside that folder along with config.yaml and samples.tsv files.
tree <path to output dir>
+
config.yaml¶
This file is used to fine tune the execution of the pipeline by setting:
- sample sheet location ... aka
samples.tsv - the temporary directory -- make sure this is correct for your computing environment.
- which circRNA finding tools to use by editing these:
- run_clear: True
- run_dcc: True
- run_mapsplice: False
- run_circRNAFinder: True
- run_nclscan: False
- run_findcirc: False
- describes the location of other resources/indexes/tools etc. Generally, these do NOT need to be changed.
samples.tsv¶
Tab delimited definition of sample sheet. The header is fixed and each row represents a sample. It has 3 columns:
- sampleName = Name of the sample. This has to be unique.
- path_to_R1_fastq = absolute path to the read1 fastq.gz file.
- path_to_R2_fastq = absolute path to the read2 fastq.gz file. If the sample was sequenced in single-end mode, then leave this blank.
The /data/CCBR_Pipeliner/testdata/circRNA/humans folder in the repo has test dataset:
tree /data/CCBR_Pipeliner/testdata/circRNA/humans
+/data/CCBR_Pipeliner/testdata/circRNA/humans
+├── GI1_N_ss.R1.fastq.gz
+├── GI1_N_ss.R2.fastq.gz
+├── GI1_T_ss.R1.fastq.gz
+└── samples.tsv
+
GI1_N is a PE sample while GI1_T is a SE sample.
Dryrun¶
Once the samples.tsv file has been edited appropriately to include the desired samples, it is a good idea to dryrun the pipeline to ensure that everything will work as desired. Dryrun can be run as follows:
charlie -w=<path to output dir> -m=dryrun
+
This will create the reference fasta and gtf file based on the selections made in the config.yaml. Hence, can take a few minutes to run.
Run¶
Upon verifying that dryrun is successful. You can then submit the job to the cluster using the following command:
charlie -w=<path to output dir> -m=run
which will produce something like this:
... ... skipping ~1000 lines
...
...
@@ -159,10 +147,10 @@
This was a dry-run (flag -n). The order of jobs does not reflect the order of execution.
Running...
14743440
-
In this example, 14743440 is the jobid returned by the slurm job scheduler on biowulf. This means that the job was successfully submitted, it will spawn off other subjobs which in-turn will be run and outputs will be moved to the results folder created inside the working directory supplied at command line. You can check the status of your queue of jobs in biowulf running:
% squeue -u `whoami`
+
In this example, 14743440 is the jobid returned by the slurm job scheduler on biowulf. This means that the job was successfully submitted, it will spawn off other subjobs which in-turn will be run and outputs will be moved to the results folder created inside the working directory supplied at command line. You can check the status of your queue of jobs in biowulf running:
squeue -u `whoami`
output:
JOBID PARTITION NAME USER ST TIME NODES NODELIST(REASON)
14743440 ccr,norm circRNA kopardev PD 0:00 1 (None)
-
ST in the above results stands for Status and PD means Pending. The status will change from pending(PD) to running(R) to completed as jobs are run on the cluster.
Next, just sit tight until the pipeline finishes. You can keep monitoring the queue as shown above. If there are no jobs running on biowulf, then your pipeline has finished (or errored out!)
Once completed the output should something like this:
% tree <path to output dir>
+
ST in the above results stands for Status and PD means Pending. The status will change from pending(PD) to running(R) to completed as jobs are run on the cluster.
Next, just sit tight until the pipeline finishes. You can keep monitoring the queue as shown above. If there are no jobs running on biowulf, then your pipeline has finished (or errored out!)
Once completed the output should something like this:
tree <path to output dir>
output:
.
├── cluster.json
├── config.yaml
@@ -856,4 +844,4 @@
├── submit_script.sbatch
└── tools.txt
Error tracking¶
Using the following command to find FAILED jobs:
grep FAIL logs/snakemake.log.jobby.short
-
The above command also gives .err and .out log files which can give further insights on reasons for failure and changes required to be made for a successful run.
Expected output:¶
The main output file is results/circRNA_master_counts.tsv.gz. Here are the top 3 tiles from an example output:
Column_number Column_title Example_1 Example_2 Example_3 1 chrom GL000220.1 GL000220.1 GL000220.1 2 start 107635 112482 118578 3 end 151634 156427 118759 4 circExplorer_strand -1 -1 -1 5 circExplorer_bwa_strand . . . 6 ciri_strand -1 -1 -1 7 dcc_strand -1 -1 -1 8 circrnafinder_strand -1 -1 -1 9 flankingsites+ CC##GC GC##CC CC##GC 10 flankingsites- GG##GC GC##GG GG##GC 11 sample_name GI1_N GI1_N GI1_N 12 ntools 1 1 1 13 HQ N N N 14 circExplorer_read_count -1 -1 -1 15 circExplorer_found_BSJcounts -1 -1 -1 16 circExplorerfound_linear_BSJ+_counts -1 -1 -1 17 circExplorerfound_linear_spliced_BSJ+_counts -1 -1 -1 18 circExplorerfound_linear_BSJ-_counts -1 -1 -1 19 circExplorerfound_linear_spliced_BSJ-_counts -1 -1 -1 20 circExplorerfound_linear_BSJ._counts -1 -1 -1 21 circExplorerfound_linear_spliced_BSJ._counts -1 -1 -1 22 ciri_read_count -1 -1 -1 23 ciri_linear_read_count -1 -1 -1 24 circExplorer_bwa_read_count 3 7 3 25 dcc_read_count -1 -1 -1 26 dcc_linear_read_count -1 -1 -1 27 circrnafinder_read_count -1 -1 -1 28 hqcounts 1 1 1 29 nonhqcounts 0 0 0 30 circExplorer_annotation Unknown Unknown Unknown 31 ciri_annotation Unknown Unknown Unknown 32 circExplorer_bwa_annotation novel novel novel 33 dcc_gene Unknown Unknown Unknown 34 dcc_junction_type Unknown Unknown Unknown 35 dcc_annotation Unknown Unknown Unknown
Expected output from the sample data is stored under .tests/expected_output.
\ No newline at end of file
+
The above command also gives .err and .out log files which can give further insights on reasons for failure and changes required to be made for a successful run.
Expected output:¶
The main output file is results/circRNA_master_counts.tsv.gz. Here are the top 3 tiles from an example output:
Column_number Column_title Example_1 Example_2 Example_3 1 chrom GL000220.1 GL000220.1 GL000220.1 2 start 107635 112482 118578 3 end 151634 156427 118759 4 circExplorer_strand -1 -1 -1 5 circExplorer_bwa_strand . . . 6 ciri_strand -1 -1 -1 7 dcc_strand -1 -1 -1 8 circrnafinder_strand -1 -1 -1 9 flankingsites+ CC##GC GC##CC CC##GC 10 flankingsites- GG##GC GC##GG GG##GC 11 sample_name GI1_N GI1_N GI1_N 12 ntools 1 1 1 13 HQ N N N 14 circExplorer_read_count -1 -1 -1 15 circExplorer_found_BSJcounts -1 -1 -1 16 circExplorerfound_linear_BSJ+_counts -1 -1 -1 17 circExplorerfound_linear_spliced_BSJ+_counts -1 -1 -1 18 circExplorerfound_linear_BSJ-_counts -1 -1 -1 19 circExplorerfound_linear_spliced_BSJ-_counts -1 -1 -1 20 circExplorerfound_linear_BSJ._counts -1 -1 -1 21 circExplorerfound_linear_spliced_BSJ._counts -1 -1 -1 22 ciri_read_count -1 -1 -1 23 ciri_linear_read_count -1 -1 -1 24 circExplorer_bwa_read_count 3 7 3 25 dcc_read_count -1 -1 -1 26 dcc_linear_read_count -1 -1 -1 27 circrnafinder_read_count -1 -1 -1 28 hqcounts 1 1 1 29 nonhqcounts 0 0 0 30 circExplorer_annotation Unknown Unknown Unknown 31 ciri_annotation Unknown Unknown Unknown 32 circExplorer_bwa_annotation novel novel novel 33 dcc_gene Unknown Unknown Unknown 34 dcc_junction_type Unknown Unknown Unknown 35 dcc_annotation Unknown Unknown Unknown
Expected output from the sample data is stored under .tests/expected_output.
\ No newline at end of file