\ No newline at end of file
diff --git a/dev/search/search_index.json b/dev/search/search_index.json
index ac715ec..a126d70 100644
--- a/dev/search/search_index.json
+++ b/dev/search/search_index.json
@@ -1 +1 @@
-{"config":{"lang":["en"],"separator":"[\\s\\-]+","pipeline":["stopWordFilter"]},"docs":[{"location":"","title":"Home","text":""},{"location":"#carlisle","title":"CARLISLE","text":"
Cut And Run anaLysIS pipeLinE
(https://CCBR.github.io/CARLISLE)
This snakemake pipeline is built to run on Biowulf.
For comments/suggestions/advice please contact CCBR_Pipeliner@mail.nih.gov.
For detailed documentation on running the pipeline view the documentation website.
The CARLISLE pipeline was developed in support of NIH Dr Vassiliki Saloura's Laboratory and Dr Javed Khan's Laboratory. It has been developed and tested solely on NIH HPC Biowulf.
"},{"location":"changelog/","title":"Changelog","text":""},{"location":"changelog/#carlisle-development-version","title":"CARLISLE development version","text":""},{"location":"changelog/#carlisle-260","title":"CARLISLE 2.6.0","text":""},{"location":"changelog/#bug-fixes","title":"Bug fixes","text":"
Bug fixes for DESeq (#127, @epehrsson)
Removes single-sample group check for DESeq.
Increases memory for DESeq.
Ensures control replicate number is an integer.
Fixes FDR cutoff misassigned to log2FC cutoff.
Fixes no_dedup variable names in library normalization scripts.
Fig bug that added nonexistent directories to the singularity bind paths. (#135, @kelly-sovacool)
Containerize rules that require R (deseq, go_enrichment, and spikein_assessment) to fix installation issues with common R library path. (#129, @kelly-sovacool)
The Rlib_dir and Rpkg_config config options have been removed as they are no longer needed.
New rules cov_correlation, homer_enrich, combine_homer, count_peaks
Add peak caller to MACS2 peak xls filename
New parameters in the config file to make certain rules optional: (#133, @kelly-sovacool)
GO enrichment is controlled by run_go_enrichment (default: false)
ROSE is controlled by run_rose (default: false)
New --singcache argument to provide a singularity cache dir location. The singularity cache dir is automatically set inside /data/$USER/ or $WORKDIR/ if --singcache is not provided. (#143, @kelly-sovacool)
contains patch for DESEQ error with non hs1 reference samples
"},{"location":"contributing/","title":"Contributing to CARLISLE","text":""},{"location":"contributing/#proposing-changes-with-issues","title":"Proposing changes with issues","text":"
If you want to make a change, it's a good idea to first open an issue and make sure someone from the team agrees that it\u2019s needed.
If you've decided to work on an issue, assign yourself to the issue so others will know you're working on it.
We use GitHub Flow as our collaboration process. Follow the steps below for detailed instructions on contributing changes to CARLISLE.
"},{"location":"contributing/#clone-the-repo","title":"Clone the repo","text":"
If you are a member of CCBR, you can clone this repository to your computer or development environment. Otherwise, you will first need to fork the repo and clone your fork. You only need to do this step once.
"},{"location":"contributing/#if-this-is-your-first-time-cloning-the-repo-you-may-need-to-install-dependencies","title":"If this is your first time cloning the repo, you may need to install dependencies","text":"
Install snakemake and singularity or docker if needed (biowulf already has these available as modules).
Install the python dependencies with pip
pip install .\n
If you're developing on biowulf, you can use our shared conda environment which already has these dependencies installed
Install pre-commit if you don't already have it. Then from the repo's root directory, run
pre-commit install\n
This will install the repo's pre-commit hooks. You'll only need to do this step the first time you clone the repo.
"},{"location":"contributing/#create-a-branch","title":"Create a branch","text":"
Create a Git branch for your pull request (PR). Give the branch a descriptive name for the changes you will make, such as iss-10 if it is for a specific issue.
# create a new branch and switch to it\ngit branch iss-10\ngit switch iss-10\n
Switched to a new branch 'iss-10'
"},{"location":"contributing/#make-your-changes","title":"Make your changes","text":"
Edit the code, write and run tests, and update the documentation as needed.
Changes to the python package code will also need unit tests to demonstrate that the changes work as intended. We write unit tests with pytest and store them in the tests/ subdirectory. Run the tests with python -m pytest.
If you change the workflow, please run the workflow with the test profile and make sure your new feature or bug fix works as intended.
If you have added a new feature or changed the API of an existing feature, you will likely need to update the documentation in docs/.
"},{"location":"contributing/#commit-and-push-your-changes","title":"Commit and push your changes","text":"
If you're not sure how often you should commit or what your commits should consist of, we recommend following the \"atomic commits\" principle where each commit contains one new feature, fix, or task. Learn more about atomic commits here: https://www.freshconsulting.com/insights/blog/atomic-commits/
First, add the files that you changed to the staging area:
git add path/to/changed/files/\n
Then make the commit. Your commit message should follow the Conventional Commits specification. Briefly, each commit should start with one of the approved types such as feat, fix, docs, etc. followed by a description of the commit. Take a look at the Conventional Commits specification for more detailed information about how to write commit messages.
git commit -m 'feat: create function for awesome feature'\n
pre-commit will enforce that your commit message and the code changes are styled correctly and will attempt to make corrections if needed.
Check for added large files..............................................Passed Fix End of Files.........................................................Passed Trim Trailing Whitespace.................................................Failed
hook id: trailing-whitespace
exit code: 1
files were modified by this hook > Fixing path/to/changed/files/file.txt > codespell................................................................Passed style-files..........................................(no files to check)Skipped readme-rmd-rendered..................................(no files to check)Skipped use-tidy-description.................................(no files to check)Skipped
In the example above, one of the hooks modified a file in the proposed commit, so the pre-commit check failed. You can run git diff to see the changes that pre-commit made and git status to see which files were modified. To proceed with the commit, re-add the modified file(s) and re-run the commit command:
git add path/to/changed/files/file.txt\ngit commit -m 'feat: create function for awesome feature'\n
This time, all the hooks either passed or were skipped (e.g. hooks that only run on R code will not run if no R files were committed). When the pre-commit check is successful, the usual commit success message will appear after the pre-commit messages showing that the commit was created.
Check for added large files..............................................Passed Fix End of Files.........................................................Passed Trim Trailing Whitespace.................................................Passed codespell................................................................Passed style-files..........................................(no files to check)Skipped readme-rmd-rendered..................................(no files to check)Skipped use-tidy-description.................................(no files to check)Skipped Conventional Commit......................................................Passed > [iss-10 9ff256e] feat: create function for awesome feature 1 file changed, 22 insertions(+), 3 deletions(-)
Finally, push your changes to GitHub:
git push\n
If this is the first time you are pushing this branch, you may have to explicitly set the upstream branch:
git push --set-upstream origin iss-10\n
Enumerating objects: 7, done. Counting objects: 100% (7/7), done. Delta compression using up to 10 threads Compressing objects: 100% (4/4), done. Writing objects: 100% (4/4), 648 bytes | 648.00 KiB/s, done. Total 4 (delta 3), reused 0 (delta 0), pack-reused 0 remote: Resolving deltas: 100% (3/3), completed with 3 local objects. remote: remote: Create a pull request for 'iss-10' on GitHub by visiting: remote: https://github.com/CCBR/CARLISLE/pull/new/iss-10 remote: To https://github.com/CCBR/CARLISLE > > [new branch] iss-10 -> iss-10 branch 'iss-10' set up to track 'origin/iss-10'.
We recommend pushing your commits often so they will be backed up on GitHub. You can view the files in your branch on GitHub at https://github.com/CCBR/CARLISLE/tree/<your-branch-name> (replace <your-branch-name> with the actual name of your branch).
"},{"location":"contributing/#create-the-pr","title":"Create the PR","text":"
Once your branch is ready, create a PR on GitHub: https://github.com/CCBR/CARLISLE/pull/new/
Select the branch you just pushed:
Edit the PR title and description. The title should briefly describe the change. Follow the comments in the template to fill out the body of the PR, and you can delete the comments (everything between <!-- and -->) as you go. Be sure to fill out the checklist, checking off items as you complete them or striking through any irrelevant items. When you're ready, click 'Create pull request' to open it.
Optionally, you can mark the PR as a draft if you're not yet ready for it to be reviewed, then change it later when you're ready.
"},{"location":"contributing/#wait-for-a-maintainer-to-review-your-pr","title":"Wait for a maintainer to review your PR","text":"
We will do our best to follow the tidyverse code review principles: https://code-review.tidyverse.org/. The reviewer may suggest that you make changes before accepting your PR in order to improve the code quality or style. If that's the case, continue to make changes in your branch and push them to GitHub, and they will appear in the PR.
Once the PR is approved, the maintainer will merge it and the issue(s) the PR links will close automatically. Congratulations and thank you for your contribution!
"},{"location":"contributing/#after-your-pr-has-been-merged","title":"After your PR has been merged","text":"
After your PR has been merged, update your local clone of the repo by switching to the main branch and pulling the latest changes:
git checkout main\ngit pull\n
It's a good idea to run git pull before creating a new branch so it will start from the most recent commits in main.
"},{"location":"contributing/#helpful-links-for-more-information","title":"Helpful links for more information","text":"
The CARLISLE github repository is stored locally, and will be used for project deployment. Multiple projects can be deployed from this one point simultaneously, without concern.
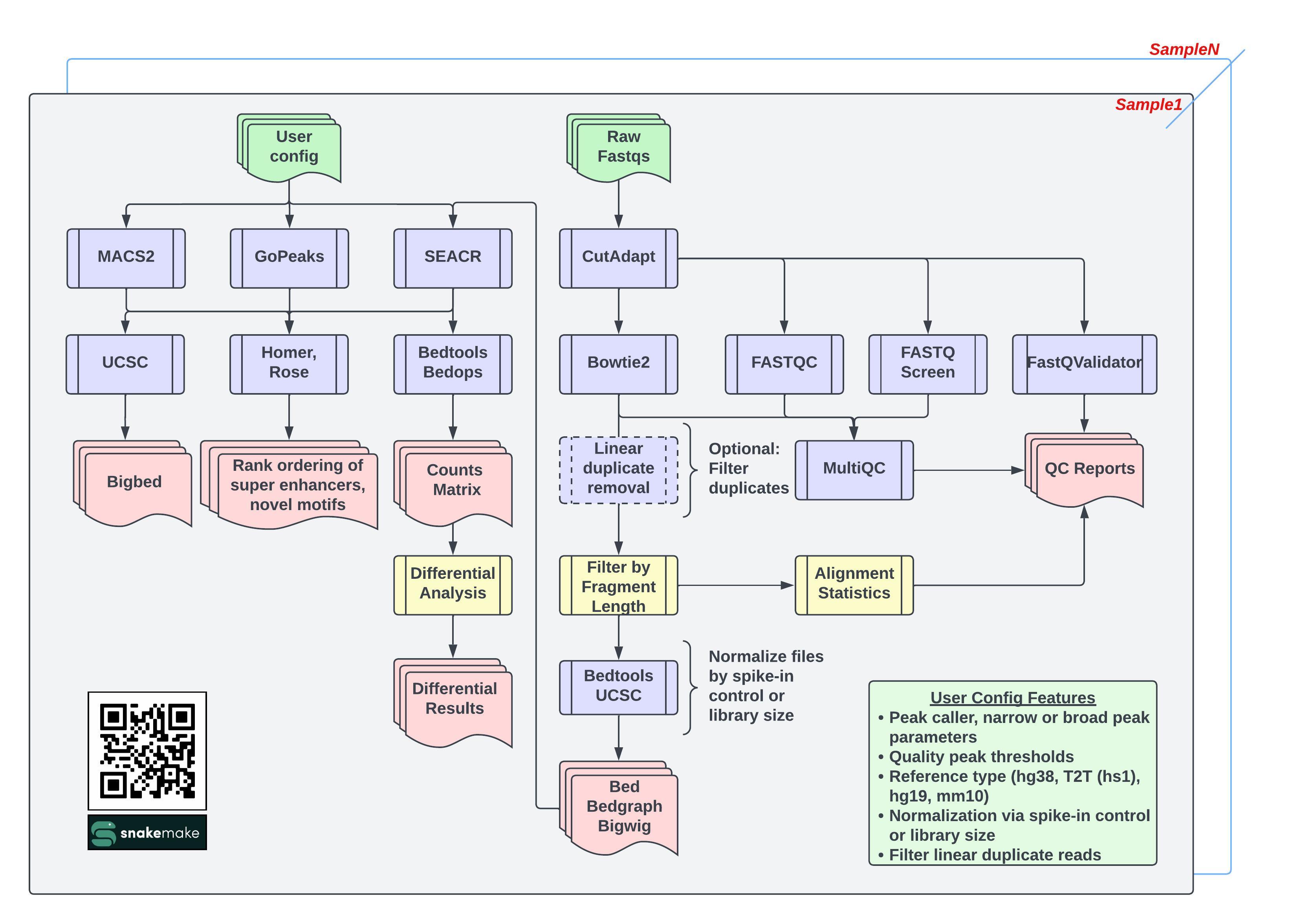
The CARLISLE Pipelie beings with raw FASTQ files and performs trimming followed by alignment using BOWTIE2. Data is then normalized through either the use of an user-species species (IE E.Coli) spike-in control or through the determined library size. Peaks are then called using MACS2, SEACR, and GoPEAKS with various options selected by the user. Peaks are then annotated, and summarized into reports. If designated, differential analysis is performed using DESEQ2. QC reports are also generated with each project using FASTQC and MULTIQC. Annotations are added using HOMER and ROSE. GSEA Enrichment analysis predictions are added using CHIPENRICH.
The following are sub-commands used within CARLISLE:
initialize: initalize the pipeline
dryrun: predict the binding of peptides to any MHC molecule
cluster: execute the pipeline on the Biowulf HPC
local: execute a local, interactive, session
git: execute GitHub actions
unlock: unlock directory
DAG: create DAG report
report: create SNAKEMAKE report
runtest: copies test manifests and files to WORKDIR
CARLISLE has several dependencies listed below. These dependencies can be installed by a sysadmin. All dependencies will be automatically loaded if running from Biowulf.
The pipeline is controlled through editing configuration and manifest files. Defaults are found in the /WORKDIR/config and /WORKDIR/manifest directories, after initialization.
The cluster configuration file dictates the resouces to be used during submission to Biowulf HPC. There are two differnt ways to control these parameters - first, to control the default settings, and second, to create or edit individual rules. These parameters should be edited with caution, after significant testing.
There are several groups of parameters that are editable for the user to control the various aspects of the pipeline. These are :
Folders and Paths
These parameters will include the input and ouput files of the pipeline, as well as list all manifest names.
User parameters
These parameters will control the pipeline features. These include thresholds and whether to perform processes.
References
These parameters will control the location of index files, spike-in references, adaptors and species calling information.
"},{"location":"user-guide/preparing-files/#2131-user-parameters","title":"2.1.3.1 User Parameters","text":""},{"location":"user-guide/preparing-files/#21311-spike-in-controls","title":"2.1.3.1.1 (Spike in Controls)","text":"
The pipeline allows for the use of a species specific spike-in control, or the use of normalization via library size. The parameter spikein_genome should be set to the species term used in spikein_reference.
Three peak callers are available for deployment within the pipeline, with different settings deployed for each caller.
MACS2 is available with two peak calling options: narrowPeak or broadPeak. NOTE: DESeq step generally fails for broadPeak; generally has too many calls.
peaktype: \"macs2_narrow, macs2_broad,\"\n
SEACR is available with four peak calling options: stringent or relaxed parameters, to be paired with \"norm\" for samples without a spike-in control and \"non\" for samples with a spikein control
peaktype: \"seacr_stringent, seacr_relaxed\"\n
GOPEAKS is available with two peak calling options: narrowpeaks or broadpeaks
peaktype: \"gopeaks_narrow, gopeaks_broad\"\n
A complete list of the available peak calling parameters and the recommended list of parameters is provided below:
Peak Caller Narrow Broad Normalized, Stringent Normalized, Relaxed Non-Normalized, Stringent Non-Normalized, Relaxed Macs2 AVAIL AVAIL NA NA NA NA SEACR NA NA AVAIL w/o SPIKEIN AVAIL w/o SPIKEIN AVAIL w/ SPIKEIN AVAIL w/ SPIKEIN GoPeaks AVAIL AVAIL NA NA NA NA
MACS2 can be run with or without the control. adding a control will increase peak specificity Selecting \"Y\" for the macs2_control will run the paired control sample provided in the sample manifest
Thresholds for quality can be controled through the quality_tresholds parameter. This must be a list of comma separated values. minimum of numeric value required.
default MACS2 qvalue is 0.05 https://manpages.ubuntu.com/manpages/xenial/man1/macs2_callpeak.1.html
default GOPEAKS pvalue is 0.05 https://github.com/maxsonBraunLab/gopeaks/blob/main/README.md
There are two manifests, one which required for all pipeliens and one that is only required if running a differential analysis. These files describe information on the samples and desired contrasts. The paths of these files are defined in the snakemake_config.yaml file. These files are:
Usage: bash ./data/CCBR_Pipeliner/Pipelines/CARLISLE/carlisle -m/--runmode=<RUNMODE> -w/--workdir=<WORKDIR>\n\n1. RUNMODE: [Type: String] Valid options:\n *) init : initialize workdir\n *) run : run with slurm\n *) reset : DELETE workdir dir and re-init it\n *) dryrun : dry run snakemake to generate DAG\n *) unlock : unlock workdir if locked by snakemake\n *) runlocal : run without submitting to sbatch\n *) runtest: run on cluster with included test dataset\n2. WORKDIR: [Type: String]: Absolute or relative path to the output folder with write permissions.\n
--help|-h : print this help. --version|-v : print the version of carlisle. --force|-f : use the force flag for snakemake to force all rules to run. --singcache|-c : singularity cache directory. Default is /data/${USER}/.singularity if available, or falls back to ${WORKDIR}/.singularity. Use this flag to specify a different singularity cache directory.
The following explains each of the command options:
Preparation Commands
init (REQUIRED): This must be performed before any Snakemake run (dry, local, cluster) can be performed. This will copy the necessary config, manifest and Snakefiles needed to run the pipeline to the provided output directory.
the -f/--force flag can be used in order to re-initialize a workdir that has already been created
dryrun (OPTIONAL): This is an optional step, to be performed before any Snakemake run (local, cluster). This will check for errors within the pipeline, and ensure that you have read/write access to the files needed to run the full pipeline.
Processing Commands
local: This will run the pipeline on a local node. NOTE: This should only be performed on an interactive node.
run: This will submit a master job to the cluster, and subsequent sub-jobs as needed to complete the workflow. An email will be sent when the pipeline begins, if there are any errors, and when it completes.
Other Commands (All optional)
unlock: This will unlock the pipeline if an error caused it to stop in the middle of a run.
runtest: This will run a test of the pipeline with test data
To run any of these commands, follow the the syntax:
Review the information on the Getting Started for a complete overview the pipeline. The tutorial below will use test data available on NIH Biowulf HPC only. All example code will assume you are running v1.0 of the pipeline, using test data available on GitHub.
A. Change working directory to the CARLISLE repository
Check your email for an email regarding pipeline failure. You will receive an email from slurm@biowulf.nih.gov with the subject: Slurm Job_id=[#] Name=CARLISLE Failed, Run time [time], FAILED, ExitCode 1
"},{"location":"user-guide/troubleshooting/#12-review-the-log-files","title":"1.2 Review the log files","text":"
Review the logs in two ways:
Review the master slurm file: This file will be found in the /path/to/results/dir/ and titled slurm-[jobid].out. Reviewing this file will tell you what rule errored, and for any local SLURM jobs, provide error details
Review the individual rule log files: After reviewing the master slurm-file, review the specific rules that failed within the /path/to/results/dir/logs/. Each rule will include a .err and .out file, with the following formatting: {rulename}.{masterjobID}.{individualruleID}.{wildcards from the rule}.{out or err}
"},{"location":"user-guide/troubleshooting/#13-restart-the-run","title":"1.3 Restart the run","text":"
After addressing the issue, unlock the output directory, perform another dry-run and check the status of the pipeline, then resubmit to the cluster.
If after troubleshooting, the error cannot be resolved, or if a bug is found, please create an issue and send and email to Samantha Chill.
"}]}
\ No newline at end of file
+{"config":{"lang":["en"],"separator":"[\\s\\-]+","pipeline":["stopWordFilter"]},"docs":[{"location":"","title":"Home","text":""},{"location":"#carlisle","title":"CARLISLE","text":"
Cut And Run anaLysIS pipeLinE
This snakemake pipeline is built to run on Biowulf.
For comments/suggestions/advice please contact CCBR_Pipeliner@mail.nih.gov.
For detailed documentation on running the pipeline, please view the website: https://CCBR.github.io/CARLISLE/
The CARLISLE pipeline was developed in support of NIH Dr Vassiliki Saloura's Laboratory and Dr Javed Khan's Laboratory. It has been developed and tested solely on NIH HPC Biowulf.
"},{"location":"changelog/","title":"Changelog","text":""},{"location":"changelog/#carlisle-development-version","title":"CARLISLE development version","text":""},{"location":"changelog/#carlisle-260","title":"CARLISLE 2.6.0","text":""},{"location":"changelog/#bug-fixes","title":"Bug fixes","text":"
Bug fixes for DESeq (#127, @epehrsson)
Removes single-sample group check for DESeq.
Increases memory for DESeq.
Ensures control replicate number is an integer.
Fixes FDR cutoff misassigned to log2FC cutoff.
Fixes no_dedup variable names in library normalization scripts.
Fig bug that added nonexistent directories to the singularity bind paths. (#135, @kelly-sovacool)
Containerize rules that require R (deseq, go_enrichment, and spikein_assessment) to fix installation issues with common R library path. (#129, @kelly-sovacool)
The Rlib_dir and Rpkg_config config options have been removed as they are no longer needed.
New rules cov_correlation, homer_enrich, combine_homer, count_peaks
Add peak caller to MACS2 peak xls filename
New parameters in the config file to make certain rules optional: (#133, @kelly-sovacool)
GO enrichment is controlled by run_go_enrichment (default: false)
ROSE is controlled by run_rose (default: false)
New --singcache argument to provide a singularity cache dir location. The singularity cache dir is automatically set inside /data/$USER/ or $WORKDIR/ if --singcache is not provided. (#143, @kelly-sovacool)
contains patch for DESEQ error with non hs1 reference samples
"},{"location":"contributing/","title":"Contributing to CARLISLE","text":""},{"location":"contributing/#proposing-changes-with-issues","title":"Proposing changes with issues","text":"
If you want to make a change, it's a good idea to first open an issue and make sure someone from the team agrees that it\u2019s needed.
If you've decided to work on an issue, assign yourself to the issue so others will know you're working on it.
We use GitHub Flow as our collaboration process. Follow the steps below for detailed instructions on contributing changes to CARLISLE.
"},{"location":"contributing/#clone-the-repo","title":"Clone the repo","text":"
If you are a member of CCBR, you can clone this repository to your computer or development environment. Otherwise, you will first need to fork the repo and clone your fork. You only need to do this step once.
"},{"location":"contributing/#if-this-is-your-first-time-cloning-the-repo-you-may-need-to-install-dependencies","title":"If this is your first time cloning the repo, you may need to install dependencies","text":"
Install snakemake and singularity or docker if needed (biowulf already has these available as modules).
Install the python dependencies with pip
pip install .\n
If you're developing on biowulf, you can use our shared conda environment which already has these dependencies installed
Install pre-commit if you don't already have it. Then from the repo's root directory, run
pre-commit install\n
This will install the repo's pre-commit hooks. You'll only need to do this step the first time you clone the repo.
"},{"location":"contributing/#create-a-branch","title":"Create a branch","text":"
Create a Git branch for your pull request (PR). Give the branch a descriptive name for the changes you will make, such as iss-10 if it is for a specific issue.
# create a new branch and switch to it\ngit branch iss-10\ngit switch iss-10\n
Switched to a new branch 'iss-10'
"},{"location":"contributing/#make-your-changes","title":"Make your changes","text":"
Edit the code, write and run tests, and update the documentation as needed.
Changes to the python package code will also need unit tests to demonstrate that the changes work as intended. We write unit tests with pytest and store them in the tests/ subdirectory. Run the tests with python -m pytest.
If you change the workflow, please run the workflow with the test profile and make sure your new feature or bug fix works as intended.
If you have added a new feature or changed the API of an existing feature, you will likely need to update the documentation in docs/.
"},{"location":"contributing/#commit-and-push-your-changes","title":"Commit and push your changes","text":"
If you're not sure how often you should commit or what your commits should consist of, we recommend following the \"atomic commits\" principle where each commit contains one new feature, fix, or task. Learn more about atomic commits here: https://www.freshconsulting.com/insights/blog/atomic-commits/
First, add the files that you changed to the staging area:
git add path/to/changed/files/\n
Then make the commit. Your commit message should follow the Conventional Commits specification. Briefly, each commit should start with one of the approved types such as feat, fix, docs, etc. followed by a description of the commit. Take a look at the Conventional Commits specification for more detailed information about how to write commit messages.
git commit -m 'feat: create function for awesome feature'\n
pre-commit will enforce that your commit message and the code changes are styled correctly and will attempt to make corrections if needed.
Check for added large files..............................................Passed Fix End of Files.........................................................Passed Trim Trailing Whitespace.................................................Failed
hook id: trailing-whitespace
exit code: 1
files were modified by this hook > Fixing path/to/changed/files/file.txt > codespell................................................................Passed style-files..........................................(no files to check)Skipped readme-rmd-rendered..................................(no files to check)Skipped use-tidy-description.................................(no files to check)Skipped
In the example above, one of the hooks modified a file in the proposed commit, so the pre-commit check failed. You can run git diff to see the changes that pre-commit made and git status to see which files were modified. To proceed with the commit, re-add the modified file(s) and re-run the commit command:
git add path/to/changed/files/file.txt\ngit commit -m 'feat: create function for awesome feature'\n
This time, all the hooks either passed or were skipped (e.g. hooks that only run on R code will not run if no R files were committed). When the pre-commit check is successful, the usual commit success message will appear after the pre-commit messages showing that the commit was created.
Check for added large files..............................................Passed Fix End of Files.........................................................Passed Trim Trailing Whitespace.................................................Passed codespell................................................................Passed style-files..........................................(no files to check)Skipped readme-rmd-rendered..................................(no files to check)Skipped use-tidy-description.................................(no files to check)Skipped Conventional Commit......................................................Passed > [iss-10 9ff256e] feat: create function for awesome feature 1 file changed, 22 insertions(+), 3 deletions(-)
Finally, push your changes to GitHub:
git push\n
If this is the first time you are pushing this branch, you may have to explicitly set the upstream branch:
git push --set-upstream origin iss-10\n
Enumerating objects: 7, done. Counting objects: 100% (7/7), done. Delta compression using up to 10 threads Compressing objects: 100% (4/4), done. Writing objects: 100% (4/4), 648 bytes | 648.00 KiB/s, done. Total 4 (delta 3), reused 0 (delta 0), pack-reused 0 remote: Resolving deltas: 100% (3/3), completed with 3 local objects. remote: remote: Create a pull request for 'iss-10' on GitHub by visiting: remote: https://github.com/CCBR/CARLISLE/pull/new/iss-10 remote: To https://github.com/CCBR/CARLISLE > > [new branch] iss-10 -> iss-10 branch 'iss-10' set up to track 'origin/iss-10'.
We recommend pushing your commits often so they will be backed up on GitHub. You can view the files in your branch on GitHub at https://github.com/CCBR/CARLISLE/tree/<your-branch-name> (replace <your-branch-name> with the actual name of your branch).
"},{"location":"contributing/#create-the-pr","title":"Create the PR","text":"
Once your branch is ready, create a PR on GitHub: https://github.com/CCBR/CARLISLE/pull/new/
Select the branch you just pushed:
Edit the PR title and description. The title should briefly describe the change. Follow the comments in the template to fill out the body of the PR, and you can delete the comments (everything between <!-- and -->) as you go. Be sure to fill out the checklist, checking off items as you complete them or striking through any irrelevant items. When you're ready, click 'Create pull request' to open it.
Optionally, you can mark the PR as a draft if you're not yet ready for it to be reviewed, then change it later when you're ready.
"},{"location":"contributing/#wait-for-a-maintainer-to-review-your-pr","title":"Wait for a maintainer to review your PR","text":"
We will do our best to follow the tidyverse code review principles: https://code-review.tidyverse.org/. The reviewer may suggest that you make changes before accepting your PR in order to improve the code quality or style. If that's the case, continue to make changes in your branch and push them to GitHub, and they will appear in the PR.
Once the PR is approved, the maintainer will merge it and the issue(s) the PR links will close automatically. Congratulations and thank you for your contribution!
"},{"location":"contributing/#after-your-pr-has-been-merged","title":"After your PR has been merged","text":"
After your PR has been merged, update your local clone of the repo by switching to the main branch and pulling the latest changes:
git checkout main\ngit pull\n
It's a good idea to run git pull before creating a new branch so it will start from the most recent commits in main.
"},{"location":"contributing/#helpful-links-for-more-information","title":"Helpful links for more information","text":"
The CARLISLE github repository is stored locally, and will be used for project deployment. Multiple projects can be deployed from this one point simultaneously, without concern.
The CARLISLE Pipelie beings with raw FASTQ files and performs trimming followed by alignment using BOWTIE2. Data is then normalized through either the use of an user-species species (IE E.Coli) spike-in control or through the determined library size. Peaks are then called using MACS2, SEACR, and GoPEAKS with various options selected by the user. Peaks are then annotated, and summarized into reports. If designated, differential analysis is performed using DESEQ2. QC reports are also generated with each project using FASTQC and MULTIQC. Annotations are added using HOMER and ROSE. GSEA Enrichment analysis predictions are added using CHIPENRICH.
The following are sub-commands used within CARLISLE:
initialize: initalize the pipeline
dryrun: predict the binding of peptides to any MHC molecule
cluster: execute the pipeline on the Biowulf HPC
local: execute a local, interactive, session
git: execute GitHub actions
unlock: unlock directory
DAG: create DAG report
report: create SNAKEMAKE report
runtest: copies test manifests and files to WORKDIR
CARLISLE has several dependencies listed below. These dependencies can be installed by a sysadmin. All dependencies will be automatically loaded if running from Biowulf.
The pipeline is controlled through editing configuration and manifest files. Defaults are found in the /WORKDIR/config and /WORKDIR/manifest directories, after initialization.
The cluster configuration file dictates the resouces to be used during submission to Biowulf HPC. There are two differnt ways to control these parameters - first, to control the default settings, and second, to create or edit individual rules. These parameters should be edited with caution, after significant testing.
There are several groups of parameters that are editable for the user to control the various aspects of the pipeline. These are :
Folders and Paths
These parameters will include the input and ouput files of the pipeline, as well as list all manifest names.
User parameters
These parameters will control the pipeline features. These include thresholds and whether to perform processes.
References
These parameters will control the location of index files, spike-in references, adaptors and species calling information.
"},{"location":"user-guide/preparing-files/#2131-user-parameters","title":"2.1.3.1 User Parameters","text":""},{"location":"user-guide/preparing-files/#21311-spike-in-controls","title":"2.1.3.1.1 (Spike in Controls)","text":"
The pipeline allows for the use of a species specific spike-in control, or the use of normalization via library size. The parameter spikein_genome should be set to the species term used in spikein_reference.
Three peak callers are available for deployment within the pipeline, with different settings deployed for each caller.
MACS2 is available with two peak calling options: narrowPeak or broadPeak. NOTE: DESeq step generally fails for broadPeak; generally has too many calls.
peaktype: \"macs2_narrow, macs2_broad,\"\n
SEACR is available with four peak calling options: stringent or relaxed parameters, to be paired with \"norm\" for samples without a spike-in control and \"non\" for samples with a spikein control
peaktype: \"seacr_stringent, seacr_relaxed\"\n
GOPEAKS is available with two peak calling options: narrowpeaks or broadpeaks
peaktype: \"gopeaks_narrow, gopeaks_broad\"\n
A complete list of the available peak calling parameters and the recommended list of parameters is provided below:
Peak Caller Narrow Broad Normalized, Stringent Normalized, Relaxed Non-Normalized, Stringent Non-Normalized, Relaxed Macs2 AVAIL AVAIL NA NA NA NA SEACR NA NA AVAIL w/o SPIKEIN AVAIL w/o SPIKEIN AVAIL w/ SPIKEIN AVAIL w/ SPIKEIN GoPeaks AVAIL AVAIL NA NA NA NA
MACS2 can be run with or without the control. adding a control will increase peak specificity Selecting \"Y\" for the macs2_control will run the paired control sample provided in the sample manifest
Thresholds for quality can be controled through the quality_tresholds parameter. This must be a list of comma separated values. minimum of numeric value required.
default MACS2 qvalue is 0.05 https://manpages.ubuntu.com/manpages/xenial/man1/macs2_callpeak.1.html
default GOPEAKS pvalue is 0.05 https://github.com/maxsonBraunLab/gopeaks/blob/main/README.md
There are two manifests, one which required for all pipeliens and one that is only required if running a differential analysis. These files describe information on the samples and desired contrasts. The paths of these files are defined in the snakemake_config.yaml file. These files are:
Usage: bash ./data/CCBR_Pipeliner/Pipelines/CARLISLE/carlisle -m/--runmode=<RUNMODE> -w/--workdir=<WORKDIR>\n\n1. RUNMODE: [Type: String] Valid options:\n *) init : initialize workdir\n *) run : run with slurm\n *) reset : DELETE workdir dir and re-init it\n *) dryrun : dry run snakemake to generate DAG\n *) unlock : unlock workdir if locked by snakemake\n *) runlocal : run without submitting to sbatch\n *) runtest: run on cluster with included test dataset\n2. WORKDIR: [Type: String]: Absolute or relative path to the output folder with write permissions.\n
--help|-h : print this help. --version|-v : print the version of carlisle. --force|-f : use the force flag for snakemake to force all rules to run. --singcache|-c : singularity cache directory. Default is /data/${USER}/.singularity if available, or falls back to ${WORKDIR}/.singularity. Use this flag to specify a different singularity cache directory.
The following explains each of the command options:
Preparation Commands
init (REQUIRED): This must be performed before any Snakemake run (dry, local, cluster) can be performed. This will copy the necessary config, manifest and Snakefiles needed to run the pipeline to the provided output directory.
the -f/--force flag can be used in order to re-initialize a workdir that has already been created
dryrun (OPTIONAL): This is an optional step, to be performed before any Snakemake run (local, cluster). This will check for errors within the pipeline, and ensure that you have read/write access to the files needed to run the full pipeline.
Processing Commands
local: This will run the pipeline on a local node. NOTE: This should only be performed on an interactive node.
run: This will submit a master job to the cluster, and subsequent sub-jobs as needed to complete the workflow. An email will be sent when the pipeline begins, if there are any errors, and when it completes.
Other Commands (All optional)
unlock: This will unlock the pipeline if an error caused it to stop in the middle of a run.
runtest: This will run a test of the pipeline with test data
To run any of these commands, follow the the syntax:
Review the information on the Getting Started for a complete overview the pipeline. The tutorial below will use test data available on NIH Biowulf HPC only. All example code will assume you are running v1.0 of the pipeline, using test data available on GitHub.
A. Change working directory to the CARLISLE repository
Check your email for an email regarding pipeline failure. You will receive an email from slurm@biowulf.nih.gov with the subject: Slurm Job_id=[#] Name=CARLISLE Failed, Run time [time], FAILED, ExitCode 1
"},{"location":"user-guide/troubleshooting/#12-review-the-log-files","title":"1.2 Review the log files","text":"
Review the logs in two ways:
Review the master slurm file: This file will be found in the /path/to/results/dir/ and titled slurm-[jobid].out. Reviewing this file will tell you what rule errored, and for any local SLURM jobs, provide error details
Review the individual rule log files: After reviewing the master slurm-file, review the specific rules that failed within the /path/to/results/dir/logs/. Each rule will include a .err and .out file, with the following formatting: {rulename}.{masterjobID}.{individualruleID}.{wildcards from the rule}.{out or err}
"},{"location":"user-guide/troubleshooting/#13-restart-the-run","title":"1.3 Restart the run","text":"
After addressing the issue, unlock the output directory, perform another dry-run and check the status of the pipeline, then resubmit to the cluster.